基于CRISPR/Cpf1系统下的诱导型多基因激活体系的建立
2021-01-28黄秋月李中瀚殷旖珂
黄秋月, 李中瀚, 殷旖珂
(四川大学生命科学学院, 成都 610064)
1 引 言
细胞的谱系分化与命运决定一直是细胞生物学研究的重点方向.体细胞重编程与功能细胞间转分化研究表明,哺乳动物细胞可通过特异性地表达少数几个特征性的转录因子而实现细胞命运的相互转化,例如Oct4、Sox2、Nanog和Lin28可将成纤维细胞重编程为诱导性多能干细胞(iPSC,induced pluripotent stem cells)[1];Ascl1、Brn2、Myt1l的表达可实现成纤维细胞向神经元转换[2];而 Hnf4α、Foxa1/2以及Gata4、Mef2c、Tbx5组合则可以分别将成纤维细胞转分化为肝实质样细胞和心肌细胞[3-4].这些研究表明,通过激活特征转录因子的表达,可实现细胞命运与功能的人工改造与转变.
传统的基因过表达策略通常依赖质粒、慢病毒、人工mRNA等形式将含有强启动子的转基因引入靶标细胞中,但是此类方法的缺陷在于当需要引入多个基因时,投递效率较低、载体能承受的基因大小受限以及外源基因整合可能破坏细胞内源基因及其调控元件等,因此,开发高效率的内源基因激活技术逐渐成为当前重编程与转分化研究中的重点方向之一.随着基因编辑技术的发展,尤其是CRISPR/Cas9系统的广泛应用,利用核酸酶活性突变体dCas9及转录激活结构域构建靶向内源基因的基因激活技术已见报道,并被成功用于重编程与转分化研究中,例如,可将MEF细胞重编程为iPSC[5]、成纤维细胞转分化为骨骼肌细胞[6]、将iPSC定向分化为神经元等[7].然而,dCas9介导的多基因激活须将多个sgRNA转录元件串联,且每个元件皆须具备U6启动子、终止子等序列,造成质粒构建繁琐且载体能承载的sgRNA元件数量有限.而近年来兴起的CRISPR/Cpf1多基因编辑因其具有RNA酶活性,能够实现pre-crRNA中多个crRNA的剪切和释放[8-9],成为更具潜力和普适性的多基因激活工具.
Cpf1蛋白晶体采用识别叶 (Recognition lobe,REC) 和核酸酶叶 (Nuclease lobe,NUC)的二裂片结构,呈蟹钳状[10].根据Cpf1识别功能域和内切酶功能域分开的特点,将NUC进行定点突变使其失去核酸内切酶活力,并与转录激活域融合构建出基因激活工具.为了更能够了解生命活动的情况,研究各生命机体生化反应下的多基因调控机制,本论文拟构建以CRISPR/Cpf1系统为基础的诱导性基因激活系统.
基于此,本论文采用了在C端融合三元转录激活域VPR(VP64-P65-Rta)的dAsCpf1、dFnCpf1、dLbCpf1,筛选获得了最强激活效果的dAsCpf1-VPR系统.此外,在该系统中还引入了不稳定结构域(DD-domain)和四环素控制的操纵子,从蛋白水平和转录水平分别控制融合蛋白表达,从而实现了特定时间、定向激活基因转录.最后,本论文实现了MYOD1、Oct4两个基因同时激活,证明了TRE3G-DD-dAsCpf1-VPR系统具有诱导性多基因激活的能力.
综上所述,在本论文中,我们成功建立了基于CRISPR/Cpf1系统的可诱导性的多基因激活体系,能够快捷、高效的通过靶向DNA进行诱导内源性激活,在转录水平进行有效调控,从而具备定向调控细胞基因表达的效果,为实现细胞命运与功能的人工改造与转变奠定技术基础.
2 材料和方法
2.1 材料和试剂
人类胚胎肾细胞系HEK 293FT,小鼠胚胎成纤维细胞系NIH3T3,DMEM basic培养基(Gibco,C11995500BT),胎牛血清(Natocor,NTC-HK009),抗生素-抗真菌剂(Gibco,15240-062),转染试剂Doge Fect (内蒙古尼迪生物,23427-01),SuperScriptTMⅡReverse Transcriptase试剂盒(Invitrogen,638315),细胞裂解液M-PER(Thermo scientific,QJ222436),GAPDH抗体(华安,R1208-3),HA抗体(Sigma,F1804-50 μg),Anti-rabbit二抗(Invitrogen,SA5-10044),Anti-mouse二抗(Invitrogen,SA5-10172),polybrene(Millipore,638313).
2.2 方 法
2.2.1 瞬时转染 6孔板中每孔铺7.5×105个细胞,24 h后,在显微镜下检查细胞覆盖率,当细胞覆盖率达到60%~70%时进行转染.转染时,取6 μL Dogo加入94 μL DMEM中涡旋混匀后,瞬离并静置5 min;同时1 μg目的质粒用DMEM补齐体积至100 μL;瞬离后加入静止5 min后的Dogo混合液涡旋混匀,室温静置30 min;最后将脂质体与质粒混合液缓慢滴入细胞孔板中,轻摇混匀后放入37 ℃、5% CO2培养箱中培养.
2.2.2 免疫印迹 去除培养基后,加入预冷的PBS,用细胞刮刀收集细胞置于1.5 mL的EP管中,3000 r/min离心5 min,去上清,加入适量细胞裂解液M-PER (Thermo scientific),用移液器吹打均匀(冰上操作).每10 min震荡一次,重复3次,随后于4 ℃,14 000 g离心20 min,收集上清,加入4×蛋白上样液,95 ℃孵育5 min使蛋白变性.SDS-PAGE电泳分离蛋白,23 V恒压半干转转膜30 min.用溶于TBST的5%脱脂奶粉封闭1 h,一抗(Anti-GAPDH 1∶2 000,Anti-HA 1∶1 000)于4 ℃冰箱中低温孵育过夜,二抗(Anti-rabbit 1∶10 000,Anti-mouse 1∶10 000)常温孵育1 h,显影.
2.2.3 荧光定量 去除培养基后,加入1×PBS清洗2~3次,Trizol法提取目的细胞的总RNA,使用Invitrogen SuperScriptTMⅡ Reverse Transcriptase试剂盒,将RNA反转录为cDNA.使用BIO-RAD SYBR Green Supermix试剂盒进行荧光定量分析,其中Gapdh为内参.
qPCR引物:
mus-Gapdh-F: GCACAGTCAAGGCCGAGAAT
mus-Gapdh-R:GCCTTCTCCATGGTGGTGAA
mus-Myod1-F:AGCGACACAGAACAGGGAAC
mus-Myod1-R:TCGAAAGGACAGTTGGGAAG
mus-Oct4-F:GCCCTCCCTACAGCAGATCACTCACATCG
mus-Oct4-R:AAGGTGTCCCTGTAGCCTCATACTCTTCTCGT
2.2.4 质粒构建
a) dAs/Fn/LbCpf1-VPR的构建
使用KOD-Plus Mutagenesis Kit(公司)点突变试剂盒分别将pcDNA3.1-As/Fn/LbCpf1-3×HA的908位、832位、917位的天冬氨酸突变为丙氨酸;在青兰基因合成VPR序列,通过infusion的方法将VPR连接在dCpf1的C端,构建成pcDNA3.1-dAs/Fn/LbCpf1-3×HA-VPR ;
b) TRE3G-DD-dAsCpf1-VPR的构建
将pLVX-TRE3G空载酶切,将DD Domain片段PCR并纯化,并将dAsCpf1-3×HA-VPR片段PCR并纯化后,用infusion的方法将DD Domian连接于dAsCpf1-3×HA-VPR的N端,构建成pLVX-TRE3G-DD-dAsCpf1-3×HA-VPR;
3 结果分析
3.1 构建CRISPR/Cpf1-VPR基因激活系统
将来源于Acidaminococcussp.的AsCpf1的第908位,Francisellanovicida的FnCpf1的第917位和Lachnospiraceaebacterium的LbCpf1的第832位的天冬氨酸突变为丙氨酸,并且在dAs/Fn/LbCpf1的C端连接了由VP64,P65和Rta(VPR)蛋白组成的三元复合转录激活因子的激活效应区域(TAD),形成具有激活作用的融合蛋白(图1a).将需激活的靶标基因的sgRNA设计在该基因转录起始位点(Transcription Start Site,TSS)之前,dCpf1-VPR在sgRNA的介导下能够特异性识别并且结合在该基因启动子或者增强子区域,VPR通过招募细胞因子或其他共转录激活因子,并结合形成多元复合物,进而定向激活靶标基因(图1b).

图1 dCpf1-VPR激活系统的构建策略
3.2 dCpf1-VPR激活系统能够定向上调基因的转录
图2为NIH3T3细胞中Oct4基因的转录水平.结果显示,dAs/Fn/LbCpf1-VPR三种激活系统都有不同程度的激活作用,其中在dAsCpf1-VPR系统下的Oct4基因mRNA水平与对照组相比可提升到104倍,证明dAsCpf1-VPR系统激活效果最强.因此我们将选用dAsCpf1-VPR激活系统进行后续的实验. 同时测试由单个sgRNA介导下的激活效果,sgRNA-2和5介导的激活效果与对照组相比提升到102倍,并且证明了由sgRNA-2和5介导的激活效果与5个sgRNA介导的的强度相同,与对照组相比可提升到104倍.
3.3 构建诱导性激活系统
为了构建诱导性基因激活系统,将TET-ON系统和不稳定配体(DD Domain)与dAsCpf1-VPR相结合,形成可由TMP和Dox小分子药物诱导的基因激活系统(图3).DD Domain是来源于大肠杆菌二氢叶酸还原酶(DHFR)突变体的不稳定结构域,能够在小分子化合物TMP的存在下保持结构稳定,促使其融合蛋白DD-dAsCpf1-VPR稳定表达.TET-ON系统中蛋白的表达受到TRE3G启动子的控制.只有在Dox存在的情况下,rtTA才能促使TRE3G启动子发挥作用并致使下游基因的转录.

图2 dCpf1-VPR系统定向激活Oct4基因转录情况

图3 构建诱导性激活系统
3.4 TRE3G-DD-dAsCpf1-VPR系统实现蛋白稳定表达和诱导性基因激活
为了验证TRE3G-DD-dAsCpf1-VPR系统的诱导控制效果,分别从蛋白水平和转录水平进行验证.图4a结果显示,在Dox和TMP双药诱导下DD-dAsCpf1-VPR融合蛋白均得到高表达,且诱导系统的本底水平表达量很低.同时图4b结果显示,加双药诱导组与对照组相比,Oct4基因的mRNA水平提高了250倍,能显著提高Oct4基因的转录水平.以上结果说明,TRE3G-DD-dAsCpf1-VPR系统可以在Dox和TMP双药的诱导下,实现对目标基因的诱导性激活.
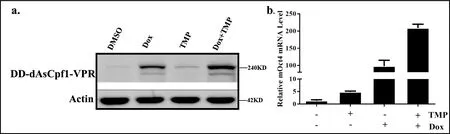
图4 Dox和TMP双药诱导蛋白表达和基因激活情况

图5 单个sgRNA介导Myod1基因的激活情况和Dox、TMP双药诱导下多基因激活的情况
3.5 TRE3G-DD-dAsCpf1-VPR系统能够介导诱导性多基因激活
图5b为Myod1基因由单个sgRNA介导的激活效果,筛选出了激活效果较强的sgRNA-1和5.然后同时对Myod1、Oct4基因进行激活,根据图5c的结果显示,Myod1、Oct4基因的转录水平分别在
Dox和TMP单药的控制下,能够实现不同程度的转录水平的提升.在Dox诱导下,Myod1和Oc4基因的转录水平分别提升了大约50倍和70倍,在同时加双药的条件下可分别提高80、120多倍,表明TRE-3G-DD-dAsCpf1-VPR系统可以高效介导诱导性多基因激活.
4 讨 论
随着基因编辑技术的发展,尤其是CRISPR/Cas9系统的广泛应用,利用核酸酶活性突变体dCas9及转录激活结构域构建靶向内源基因的基因激活技术已见报道,并被成功用于重编程与转分化研究中.然而,dCas9介导的多基因激活须将多个sgRNA转录元件串联,且每个元件皆须具备U6启动子、终止子等序列,造成质粒构建繁琐且载体能承载的sgRNA元件数量有限.CRISPR/Cpf1系统是近年来兴起的多基因编辑系统,其突出特点是,Cpf1具有RNA酶活性,能够促使pre-crRNA中的多个crRNA的剪切和释放,因此更具备作为多基因激活工具的潜力和普适性.
为了能够更好地实现细胞命运与功能的人工改造与转变,本文构建了基于CRISPR/Cpf1系统下能够定时定向激活多基因的工具.为了筛选出最高效激活能力的Cpf1,对比了dAs/Fn/LbCpf1-VPR的激活效果.基于此,为了构建可诱导的Cpf1基因激活技术方法,在dAsCpf1-VPR的N端引入了不稳定结构域(DD-domain)和四环素控制的操纵子,通过在转录和蛋白稳定性两个层面添加诱导性调控元件,证明能够在Dox和TMP双药调控下蛋白水平和转录水平的高效诱导表达和激活.最后,对MYOD1、Oct4两个基因同时激活,证明TRE3G-DD-dAsCpf1-VPR系统具有诱导性多基因激活的能力.综上所述,成功将CRISPR/Cpf1系统与TET-ON系统融合,建立了诱导性多基因激活体系,能够快捷、高效的通过靶向DNA进行诱导内源性激活,在DNA水平上对细胞转录水平进行有效调控,具有定向调控细胞基因表达,能够实现细胞命运与功能的人工改造与转变的潜力.
