Genome-wide Association Analysis of Maize Flowering Traits
2021-01-26HaiyingZHANGShuGAOBinyangLIHaixuZHONGZhichengZHANGBowenLUO
Haiying ZHANG, Shu GAO, Binyang LI, Haixu ZHONG, Zhicheng ZHANG, Bowen LUO,2*
1. Maize Research Institute, Sichuan Agricultural University, Chengdu 611130, China; 2. State Key Laboratory of Crop Gene Exploration and Utilization in Southwest China, Chengdu 611130, China; 3. College of Agronomy, Sichuan Agricultural University, Chengdu 611130, China
Abstract Flowering regulation is important for maize to adapt to a variety of environments as well as associated with high yield. In this study, the genetic mechanism of three flowering traits of 310 maize inbred lines with rich genetic background was investigated in three years at three different environments such as days to tasseling (DTT), days to silking (DTS) and days to pollen shedding (DTP). Based on mean performance, the longest flowering time was observed in Zhanyi (2018), whereas the shortest in Shizong (2019). The coefficient of variance depicted the range from 3.62% to 9.06% for three flowering traits under all environments. Therefore, we have integrated these flowering traits corresponding to SNP molecular markers for genome-wide association study (GWAS). Results showed that 22 SNPs markers were significantly associated with DTT according to physical position and average linkage disequilibrium (LD) decay distance, and a total of 234 candidate genes were identified near these significantly associated SNP markers. Moreover, KEGG and GO analysis showed that these genes were enriched in the regulation of the physiological pathways for flowering. In more details, 16 genes involved in development of floral organs are more worthy of our attention in future studies.
Key words Maize, Flowering trait, Genome-wide association analysis (GWAS)
1 Introduction
The flowering time reflects the transitional process of vegetative growth to reproductive growth in the life cycle of maize. It is closely related to the regional and seasonal adaptability of maize varieties and affects the seed setting rate of maize ears[1]. Therefore, understanding the genetic mechanisms of maize flowering traits not only provides useful information for maize adaptability improvement and high yield breeding, but also lays the foundation for further fundamentally molecular research.
Recently, the QTL mapping of flowering traits has been reported by an increasing number of studies[2]. Sunetal.[3]identified the 11 QTLs associated with a flowering time from the 188 F2∶3 population. Meanwhile, Agramaetal.[4]found 3 ASL (interval between anthesis)-associated QTLs using the maize F3 population. Buckler’s study[5]showed that the difference of flowering time was not caused by the QTLs with large effects, but rather by countless small-effect QTLs. Besides, Chardonetal.[6]integrated a large number of QTLs from different mapping populations to perform meta-analysis, and the hot regions of consistent QTLs on chromosome 1, 8, 9, and 10 were identified. Because QTL mapping uses man-made recombination events, the number of recombination is limited due to the limitation of hybridization and selfing, so the accuracy of QTL mapping is low and can only be located in a large range. It is necessary to create near-isogenic lines in order to locate candidate genes.
Another kind of mapping method is association mapping, also known as linkage disequilibrium mapping and it is a relatively new and promising method for the mapping of complex quantitative traits. Based on the concept of linkage disequilibrium, association analysis uses historical recombination events accumulated around the target traits and natural genetic diversity within the population for gene mapping, which has a high mapping accuracy. Some GWAS studies related to flowering have been reported in maize. For example, a total of 26 SNPs which were significantly associated with tassel number were detected by genome-wide associated analysis (GWAS)[7]. In addition, 106 selective-sweep regions containing 423 candidate genes were revealed by selective signature analysis and SNP- and haplotype-based GWAS were performed with 39 350 high-quality SNP markers[8]. To detect more variations associated with maize flowering time, Lietal.[9]used a population containing more than 8 000 lines and nearly 1 million single nucleotide polymorphisms (SNPs) to conduct GWAS and identified about 220 candidate genes in 90 genomic regions. Although there have been so many studies on the mapping of maize flowering, only a few genes related to flowering have been cloned, such asVgt1[10],ZmCCT[11]andNut1[12]. So, the molecular mechanisms and regulatory pathways for flowering time in maize need to be further explored.
In this study, we investigated the days to tasseling (DTT), days to silking (DTS) and days to pollen shedding (DTP) of 310 genotypically diverse inbred lines from the Southwest China breeding program at three environments in three years. GWAS was conducted using the 46 209 high-quality SNPs and BLUP values of three flowering traits of the 310 inbred lines to screen flowering associated candidate genes. It will provide stable mapping results for further understanding the regulation of the maize flowering.
2 Materials and methods
2.1PlantmaterialsA total of 310 genotypically diverse inbred lines from the current Southwest China breeding program were screened to investigate flowering traits. The field experiments were conducted during the natural growing season in 2018-2020 at three experimental locations, Zhanyi (Yunnan Province, 2018; southwest region), Jiuquan (Gansu Province, 2018 and 2019, northwest region) and Shizong (Yunnan Province, 2019, southwest region), with different hours of sunlight. The tested materials were sowed on March 3, May 30, and May 3 at each environmental location, respectively. The plots were arranged in a complete randomized block design of single row plots with two replications at each location. Each row plot was 3 m in length with 0.75 m spacing between the rows and 0.3 m between plants.
2.2PhenotypicidentificationanddataanalysisDays to tasseling (DTT), days to silking (DTS), and days to pollen shedding (DTP) were investigated for each plot as the interval from the date of sowing to the date on which 50% of the plants in the plot had initiated tasseling, silking, and pollen shedding, respectively. Descriptive analysis and analysis of variance (ANOVA) were calculated by R software 3.3.3. After eliminating the abnormal values of each phenotype, the best linear unbiased predictive value (BLUP) of each flowering trait under different environmental condition was calculated using the lme4 package in R software 3.3.3[7]. The broad-sense heritability formula for each phenotype was performed using the model as follows[13]:
h2=σg2/σg2+σge2+σe2
where σg2 is the variance of the genotype, σge2 is the variance of the interaction between genotype and environment, and σe2 is error variance.
2.3Genome-wideassociationanalysisandcandidategenescreeningThe 46 209 high-quality genomic SNPs of the 310 maize inbred lines (with missing rate <20% and minor allele frequency >0.05) were used for GWAS[14]. Combined with the BLUP value of the flowering traits, the GWAS was calculated by using the MLM model (Q+Kmodel) in Tassel 5.2.30[15]. The results of the population structure (Q) and the kinship matrix (K) were estimated from a previous study[14]. For 46 209 markers, the corresponding Bonferroni-corrected thresholdPvalues at α=1 and α=0.05 were 2.16×10-5and 1.08×10-6(logPvalues of 4.66 and 5.97, respectively). So, we setP<2.16×10-5as the standard to determine whether an SNP locus was significantly associated with a trait. According to the average LD decay distance, we extracted the genes near the significant association sites from the public maize genome data set (www.maizegdb.org). GO and KEGG-pathway enrichment analysis of these candidate genes were performed by ClueGo[16].
3 Results and analysis
3.1PhenotypicanalysisbasedonthreeenvironmentalconditionsDTT, DTS, and DTP of 310 maize inbred lines in three environmental conditions all varied widely (Table 1). The mean of DTT ranged from 47.49 to 110.68 d. The mean of DTS ranged from 51.56 to 114.73 d. And the mean of DTP ranged from 49.74 to 113.75 d. It suggested that the maize germplasm contained abundant genetic variations in flowering traits. Among different environments, the flowering time at Zhanyi in 2018 was the longest, with an average DTT of 110.68 d, DTS of 114.73 d, and DTP of 113.75 d, and the corresponding coefficients of variation ranged from 3.62% to 4.53%. Whereas the flowering time at Shizong in 2019 was the shortest, with an average DTT of 47.49 d, DTS of 51.16 d, DTP of 49.74 d, and the coefficients of variation ranged from 6.64% to 9.06%.
3.2MiningcandidategenesrelatedtofloweringbyGWAS
A total of 22 single-nucleotide polymorphism (SNP) markers were significantly associated with DTT and the contribution rate ranged from 5.948% to 19.361% (Table 2). Among the 22 SNPs, 13 SNP markers were extremely significantly associated with DTT. According to the physical position of these SNP markers and average LD (linkage disequilibrium) decay distance, 234 candidate genes were identified near these significantly associated SNP markers. These candidate genes were mainly distributed on chromosome 1, 4, with few on chromosome 3, 9. The GO and KEGG pathway enrichment of these 234 candidate genes were analyzed by ClueGO, the results showed that these genes were mainly involved in meiosis, synthesis of plasma membrane protein complexes, and plant-type vacuoles. In more detail, 16 genes were annotated to be involved in autonomous, photoperiod, pollen germination pathways,etal.
The functions of the 16 corresponding homologous genes all have been verified inArabidopsis(Table.3). According to the function and annotation of homologous genes, we divided these 16 candidate genes into four categories. Firstly, 11 homologous genes involved in the development of pollen germination and pollen tube growth were obtained, including 7 genes screened from transcriptome analysis[17-19]. Then, some studies reported that EMF1 and BAS1 are involved in photoperiod or photomorphogenesis[20-21]. Thirdly, ATLP-3 was identified by genome-wide analysis of spatial gene expression inArabidopsisflowers[22]. Besides, as a metallo-phosphoesterase, the expression of ATPAP3 was detected in specific flower organs[23]. Finally, SDS encodes a meiotic cyclin-like protein that is necessary for meiotic DNA double-strand breaks (DSBs) repair and it was also reported to affect the fertility[24].
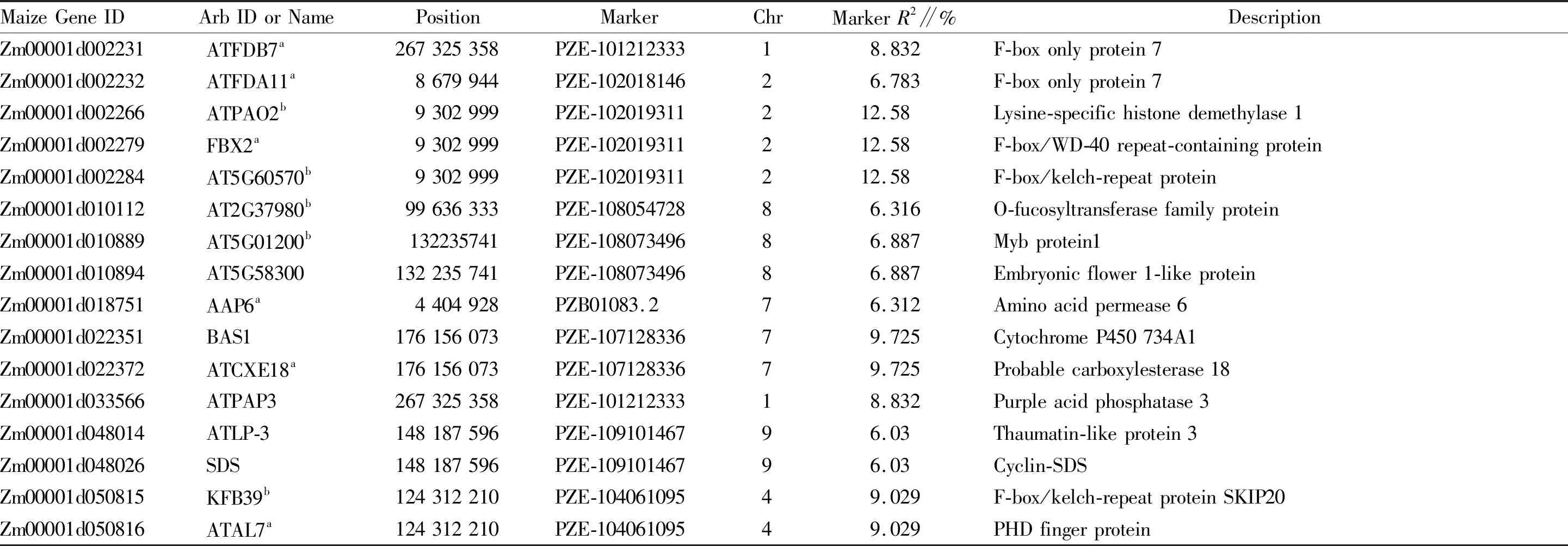
Table 3 The 16 genes involved in flowering among the 234 candidate genes
4 Discussion
In this study, three environmental locations were selected in northern and southern regions, namely, the areas of long-day and short-day respectively, which have a great impact on the growth of maize. The phenotypic values with extremely large changes in flowering time under different environmental conditions were found in the experimental results, especially some photoperiod-sensitive materials, indicating that the adaptability of maize to the environment is still different. Although the field experiment is easily affected by environmental factors, it is more suitable for us to screen extreme materials.
GWAS is a powerful method for analyzing complex quantitative traits, it uses molecular markers distributed throughout the genome to associate with target traits[8]. In our study, only the tasseling trait was significantly associated with SNPs. It may be caused by the relatively lower density of SNPs which does not completely cover the genes that control other flowering traits. In this study, the flowering traits were recorded under multiple environment conditions, and the BULP values were estimated for each line across the different environments, which can help to identify stable genetic locus[7]. Maize flowering, pollen germination, and pollen tube elongation are a series of complex biological processes, involving in many signal transduction and physiological metabolic pathways. The analysis of GO and KEGG pathway enrichment showed that most of the candidate genes were mainly involved in meiosis. As an important process of gametes producing (eggs and germs) at flowering stage, meiosis plays a very important role in the seed setting rate of plants[25]. This result is a good illustration of the accuracy of the flowering related candidate genes screened by GWAS.
In addition, the homologs of 16 candidate genes screened from the GWAS results were verified inArabidopsisthaliana. In the process of pollen germination (PG) and pollen tube growth (PTG),ATFDB7,ATFDA11 andATAL7 will generate proteins via intercompartmental duplication in anthers and pollen organellar to fight against microbial pathogens[18]. Meanwhile, the transcriptome analysis showed that seven genes (ATPAO2,FBX2,AT5G60570,AT2G37980,AT5G01200,ATLP-3,KFB39) have initiated transcription to mobilize materials and energy for PG with PTG that increase the physiological and biochemical activities of germinating pollen and pollen tubes[17]. Besides, AAP6 was interpreted to be a higher affinity system that can transfer acidic and neutral amino acids as the nitrogen source to the locule for pollen development[19]. In the signal response of photoperiod and photomorphogenesis, EMF1 can integrate LHP1 and a trimethyl histone H3lysine-4 (H3K4) demethylase to form a complex, which can silence the florigen geneFLOWERINGLOCUST(FT) to prevent photoperiod-independent flowering and make flowering time respond to environmental signals in Arabidopsis[20]. Furthermore, the photomorphogenesis could occur at multiple stages of plant growth and development, such as the flowering time. After floral induction, the expression ofBAS1 was observed throughout the shoot apex that may prevent an early flowering transition by transporting brassinosteroids[21]. In flower organs,ATPAP3 has been identified as a metallo-phosphoesterases that could activate the process of dephosphorylation and was found to express specifically in some flower organs, indicating that it may be related to the flowering of plants[23]. Meanwhile,ATLP-3 expressed in tapetum of anthers encodes a thaumatin-like protein that can accelerate the cellular differentiation in petals and stamens[22]. Lastly,SDSis a meiosis specific cyclin protein involved in meiosis. In order to enhance the fertility of male gametes,SDScan act withASY1 andAtHOP2/AHP2 to drive DNA double-strand breaks repair[24].
Although these candidate genes have not been verified in maize, the verification results of the homologs of these genes in other model plants provide us important reference information and increase the reliability of our screened candidate genes. These results will provide valuable information for further study of maize flowering regulation mechanisms in the future.
杂志排行

Asian Agricultural Research的其它文章
- Practice of Mind Mapping Technology in "Re-flipped Classroom" Teaching Mode: A Case Study of the Course Principles of Residential District Planning
- A New Record of Orchidaceae from Guangxi: Galeola nudifolia Lour.
- Quality Evaluation of Threshing and Redrying Process Based on Analytic Hierarchy Process
- Development Strategy of Soundscape in Rural Tourism Based on Tourist Perception: A Case Study of Some Villages in Jiangxi Province
- Sustainable Utilization of Agricultural Land in Chengdu Metropolitan Area Based on Emergy Analysis
- Effect of Different Processing Methods on Storage Quality of Strawberries