Discovery of human coronaviruses pan-papain-like protease inhibitors using computational approaches
2021-01-21MubrkAlmriMuhmmdThirulQmrMuhmmdUsmnMirzSfrAlqhtniMtheusFroeyenLingLingChen
Mubrk A.Almri,Muhmmd Thir ul Qmr,Muhmmd Usmn Mirz,Sfr M.Alqhtni,Mtheus Froeyen,Ling-Ling Chen
aDepartment of Pharmaceutical Chemistry,College of Pharmacy,Prince Sattam Bin Abdulaziz University,Alkarj,Saudi Arabia
bCollege of Life Science and Technology,Guangxi University,Nanning,China
cDepartment of Pharmaceutical and Pharmacological Sciences,Rega Institute for Medical Research,Medicinal Chemistry,University of Leuven,B-3000,Leuven,Belgium
dHubei Key Laboratory of Agricultural Bioinformatics,College of Informatics,Huazhong Agricultural University,Wuhan,430070,China
ABSTRACT
Keywords:
COVID-19
MERS-CoV
Molecular dynamic simulation
Pan-inhibitors
Papain-like protease
SARS-CoV
SARS-CoV-2
Virtual screening
1.Introduction
Coronaviruses(CoVs)are enveloped RNA viruses covered with a non-segmented positive-sense RNA genome of 28-30 kb,known since the mid-1960s[1].These viruses can infect a variety of hosts and can cause different respiratory,enteric,liver,and systemic diseases[1,2].CoVs have the potential to transmit among animals and humans[3,4],which is evident from previous CoVs outbreaks.The severe acute respiratory syndrome(SARS)surfaced in 2002/2003 and resulted in 800 deaths[5].Soon after the rise of this new viral infection,several new CoVs were uncovered[6].In 2012,Middle East respiratory syndrome coronavirus(MERS-CoV)was identified with the potential of human-to-human transmission[7,8].The mortality rate of SARS-CoV and MERS-CoV was estimated to be around 15% for SARS-CoV[9]and 35% for MERS-CoV[10].The currently emerged SARS-CoV-2 spread globally and turned into the pandemic of life threatening coronavirus disease-2019(COVID-19)[11].To date,various potential SARS-CoV-2 drug-targets have been reported,and various efforts have been made to identify potential therapeutics as presented in recent reviews[11-15].
SARS-CoV-2 belongs to beta-CoVs,and is based on 800-kDa polypeptide,which is cleaved into structural and non-structural proteins upon translation[16,17].3-chymotrypsin-like protease(3CLpro)and papain-like protease(PLpro)are active partners in mediating this proteolytic processing.CoVs PLprois grouped in the peptidase clan CA(family C16).The active site of PLproconsists of a typical catalytic triad,comprising Cys111-His272-Asp286 residues.CoVs PLprois extensively studied,well-aligned,functional,and situated at the border of the thumb and palm sub-domains[18].PLproperforms its proteolytic functions through its catalytic cysteineprotease cycle,in which Cys111 functions as a nucleophile,His272 acts as a general acid/base and Asp286 is linked with the histidine assisting it to align and help deprotonation of Cys111[19].CoVs 3CLproand PLpromainly process the viral polyprotein.However,PLprohas an extra role of extracting ISG15 and ubiquitin from the proteins of host-cell to help CoVs in the dodging of host-innate immune responses[19,20].The C-terminal of ubiquitin molecule is suggested to accommodate a cleft in close proximity to the functional catalytic triad which consists of the conserved ubiquitin-specific protease residue,Asp 164,and two hydrophobic subsites S3 and S4.Targeting this pocket is preferable for the development of non-covalent SARSCoV agents as it could allosterically block the active site by inducing loop closure[21].Therefore,PLprois a significant target for anti-CoVs drug designing[22].PLprobased antiviral drugs may not only inhibit the CoVs replication cycle;they may also have an advantage in impeding the dysregulation of signalling cascades in CoVs infected cells,further leading to the death of un-infected neighbouring cells[19].
Up to date,there is no clinically approved drug or vaccine available to protect against recent COVID-19[23,24].For treating COVID-19 pneumonia,health officials are currently testing and evaluating existing anti-pneumonia treatments.Existing antivirals,including protease inhibitors(indinavir,saquinavir,and lopinavir/ritonavir),as well as RNA polymerase inhibitors,including remdesivir[25-27],are being tested against SARS-CoV-2.Recently,the in vitro antiviral competence analyses of a few FDA-approved as well as experimental drugs against clinical isolates of SARS-CoV-2,such as chloroquine and remdesivir,showed promising results[26].However,to tackle the current COVID-19 pandemic,the development of wide-spectrum inhibitors against CoVs is a crucial strategy.
In recent years,the use of computational approaches for the discovery of small molecules has achieved importance in drug development[28-33].Among various approaches,molecular docking has been extensively used for investigating the binding interactions of small molecules with the active sites of the target protein[34-38].In antiviral drug discovery,hierarchical virtual screening approaches have already identified promising antiviral compounds against a broad range of viruses including influenza[39],Ebola[40-42],Dengue[43-48],Zika Virus[48-50]and recently against CoVs[22,51-53],while others display the significance of molecular dynamics(MD)simulations in search for possible antiviral[44,54-61]and investigated drug resistance mechanisms[54,58,62-64].PLprois a highly conserved protease across CoVs[65]and considered as a potential target for SARS-CoV inhibitors with broad-spectrum antiviral activity[19].In this contribution,a combined virtual screening approach and all-atom MD simulations were employed to investigate potential pan-PLproinhibitors that could be further developed into broad-spectrum anti-COVID-19 drugs.
2.Materials and methods
2.1.Proteins sequence and structure alignment
The functional evolutionary conserved residues of SARS-CoV,SARS-CoV-2 and MERS-CoV were recognized through sequence and structure alignments that could provide a structural motif for inhibitor design towards the discovery of pan-PLprobased broad spectrum anti-COVID-19 hits.Sequence and 3D structures of SARSCoV-2 PLpro(Protein Data Bank(PDB):6W9C),SARS-CoV PLpro(PDB:3MJ5),and MERS-CoV PLpro(PDB:4R3D)were retrieved from PDB[66].Clustal Omega was used to align the PLprosequences[67].Structure alignment analysis was performed using PyMOL 1.3.tool[68].
2.2.Chemical libraries preparation
The Asinex protease inhibitor library composed of 6,968 compounds in three-dimensional(3D)representation and structural data format(SDF)was downloaded from the Asinex platform(https://www.asinex.com/protease/).The compounds were imported,and energy minimized using MMFF94 force field implemented in Open Babel[69].Then compounds were prepared for screening using Autodock Tools[70]by adding the polar hydrogens and computing the gasteiger charges.All the optimized compounds were then saved as PDBQT files format for further molecular docking studies.
2.3.Structure-based virtual screening and molecular docking
The x-ray structure of SARS-CoV-2 PLproin a resolution of 2.7 Å(PDB:6W9C)was used for the docking purpose.Initially,the cocrystalized inhibitors and unwanted water molecules were removed by the Discovery Studio Visualizer[71].The protein structure was prepared from PDB files into PDBQT using Autodock Tools.The polar-hydrogen atoms were included,and gasteiger charges were processed before docking.The structure-based virtual screening was carried out using Autodock-vina in PyRx 0.8 virtual screening tool[72].Due to the high degree of sequence identity,the structure of SARS-CoV PLpro(PDB:3MJ5)bound to GRL-0667S,a non-covalent inhibitor,was used to determine the target site within SARS-CoV-2 PLpro.The docking target site was determined by 3D structural alignment of SARS-CoV-2 PLpro(PDB:6W9C)with SARS-CoV PLpro(PDB:3MJ5).Hence,the grid box was generated by confining the essential residues lining this binding cavity.The three top hits were then re-docked individually using Autodock-Vina 1.1.2 to predict their binding modes and mechanism of interactions.The docking parameters were initially validated by redocking of native ligand GRL-0667S into the active site of SARS-CoV PLpro(PDB:3MJ5).Also,these hits were docked against SARS-CoV PLpro(PDB:3MJ5)and MERS-CoV PLpro(PDB:4R3D)using the same method.Discovery Studio Visualizer 4.5[73]and PyMOL 1.3[68]programs were used for data and interaction analyses.
2.4.In silico drug-likeness analysis and ADMET profiling
The available bioinformatics tool SwissADME(available online:http://www.swissadme.ch/index.php)[74]was used for finding the drug-likeness properties.Based on the Lipinski's rule of five[75],the properties that have been considered were molecular mass(MW),H-bond donor(HBD),H-bond acceptor(HBA),lipophilicity(log P),aqueous solubility and rotatable bonds(QP log S).PreADMET(https://preadmet.bmdrc.kr/)and pkCSM (http://biosig.unimelb.edu.au/pkcsm/)[76]servers were used to determine the ADMET(absorption,distribution,metabolism,excretion,and toxicity)parameters of candidate compounds.
2.5.MD simulation
All atoms MD simulation of SARS-CoV-2 PLpro-inhibitor complexes and apo-protein were performed at 50 ns using GROMACS 2018 package[77].The simulation was carried out using previously reported protocol[53,78].Briefly,UCSF Chimera 1.14 was used to prepare the crystal structure of apo-SARS-CoV-2 PLproand in complex with the top pose of docked-compounds for MD simulation[79].The topology and parameters of compounds were obtained using SwissParam (http://www.swissparam.ch/).The simulation was conducted by applying OPLS-AA/L force-field to the systems in a 3D cubic box of TIP3P model of water molecules.Next,the simulated systems were equilibrated,and energy minimized by steepest-decent algorithm,followed by equilibration using Canonical(NVT)as well as(isothermal/isobaric)NPT ensembles.The MD simulation was analyzed for the root-mean square deviation(RMSD),root-mean square fluctuations(RMSF),potential binding energy,a radius of gyration(Rg),H-bond interaction analysis,solvent accessible surface area(SASA)and principal component analysis(PCA).
2.6.Binding free-energy calculations
The binding free-energies(ΔGbind)of the candidates were computed using the MM-PBSA algorithm[80],employed in AMBER 18,as previously described[81,82].The molecular mechanics(MM)force fields were utilized to calculate the energy contributions of the receptor,ligand,and complex in a gaseous phase.The total binding free-energy(ΔGtotal)is determined as a total energy released from the ligand/protein complex which is contributed by molecular mechanics binding energy(ΔEMM)and solvation free energy(ΔGsol)using the following equations:

In which,ΔEMMis divided into internal energy(ΔEint),electrostatic energy(ΔEele),and van der Waals energy(ΔEvdw),and the polar(ΔGp)and non-polar(ΔGnp)energy components contributed to total solvation free energy(ΔGsol).ΔGbindis the free energy of binding evaluated after entropic calculations(-TΔS),for both MMGBSA and MM-PBSA methods.In order to estimate the decisive role of interacting residues towards ligand's binding,per-residue energy decomposition analysis was performed using the MM-GBSA method,and binding energy was calculated as ΔGresidueusing the following equation.
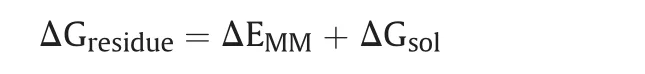
The ΔGresidueincludes the total energy obtained from sidechain and backbone energy decomposition.These methods have been well demonstrated in binding free energy calculations[83]for antiviral inhibitors[84,85].
3.Results and discussion
3.1.Sequence,structural and functional analysis of PLprofor conserveness among coronaviruses
The sequence alignment of SARS-CoV-2 PLprodisplayed an identity of 82.80% and only 30.00% with SARS-CoV PLproand MERSCoV PLpro,respectively(Fig.1).However,the sequence alignment revealed that PLprocrucial catalytic triad residues of CoVs PLprowere well conserved amongst SARS-CoV-2 (Cys111-His272-Asp286),SARS-CoV (Cys112-His273-Asp287)and MERS-CoV(Cys112-His275-Asp294).
In consistence with this,the structural alignment/superposition of all three human CoVs PLprorevealed that the PLproof SARS-CoV-2(Fig.2A)adapted the same folding pattern as the PLproof SARS-CoV and MERS-CoV(Fig.2B).Interestingly,the functionally wellconserved catalytic triad residues within the catalytic pockets of PLproamong SARS-CoV-2,SARS-CoV,and MERS-CoV present at the identical place in the catalytic sites with RMSD 1.342 Å as presented in Figs.2B and C.To determine the targetable binding site within the SARS-CoV-2 PLpro,the crystal structure of SARS-CoV PLproin complex with a potent non-covalent inhibitor,GRL-0667S,was used for structural alignment.The binding site appeared as an allosteric site close to the active catalytic site(Figs.2B and D).
3.2.Virtual screening of protease inhibitor library
The integrated computational method comprising virtual high throughput screening,molecular docking,and MD simulation is a significant approach for the exploration of potential inhibitors against a target protein[22,28,78,86].In order to find out potential pan-PLprobased anti-SARS-CoV-2 inhibitors,the structure-based screening was carried out against a virtual library of~7,000 protease inhibitors.By applying a docking score cutoff of lower than-8.5 kcal/mol,three potential hits(Fig.3)were selected with maximum scores,which were found to interact well with the active site residues.These include ADM_13083841((S)-4-(2-(2-(5-methyl-7-oxo-6,7-dihydropyrazolo[1,5-a]pyrimidin-2-yl)pyrrolidin-1-yl)-2-oxoethyl)phthalazin-1(2H)-one),AEM_16392818LMG_15521745(N-(2-(3H-pyrazol-4-yl)ethyl)-4-((3-(2-fluorophenyl)isoxazol-5-yl)methyl)tetrahydro-2H-pyran-4-carboxamide)and SYN_15517940((R)-2-methyl-6-(((1R,5R)-8-oxo-1,5,6,8-tetrahydro-2H-1,5-methanopyrido[1,2-a][1,5]diazocin-3(4H)-yl)sulfonyl)-2H-benzo[b][1,4]oxazin-3(4H)-one),with binding energy score of-8.9,-8.7,-8.7 kcal/mol,respectively,and considered as potential inhibitors of SARS-CoV-2 PLpro(Table 1).These three hit compounds were used to further evaluate the physiochemical and ADMET properties.
3.3.Analysis of screened inhibitor interaction
In order to understand the binding mode and mechanism of interaction of these compounds with SARS-CoV-2 PLpro,an unbiased flexible docking of these compounds into the active site of the SARS-CoV-2 PLproenzyme was performed.The docking and scoring functions had been validated before the docking was carried out.Since SARS-CoV and SARS-CoV-2 shared significant sequence similarity and similar 3D structure,the validation was achieved by docking the GRL-0667S,a potent SARS CoV PLproinhibitor with an IC50value of 0.32±0.01μM,into the same site within SARS-CoV-2[87].The latter approach was made to evaluate the ability of the docking protocol to predict the biologically active conformation.Moreover,as shown in Fig.4,both the docked conformation within SARS-CoV-2 and co-crystal ligand within SARS-CoV adapted a similar binding mode within the target site,validating the robustness of the docking protocol.
Flexible docking revealed that the three compounds adopted the same binding mode within the binding pocket and interacted through H-bonds as shown in Fig.5.Among the conserved H-bonds interactions,the terminal pyrimidin-4-one of ADM_13083841,central oxane moiety of LMG_15521745 and terminal benzoxazines moiety of SYN_15517940 established one H-bond each with the side chain oxygen atom of Thr301.The second conserved H-bond was established between the side chain oxygen of Tyr264 and the central pyrrolidine moiety of ADM_13083841,oxazole moiety of LMG_15521745,and sulfonamide moiety of SYN_15517940.Apart from these,ADM_13083841 and LMG_15521745 also established conserved interactions with the side-chain nitrogen(N)of Lys157,while SYN_15517940 established H-bond with the side-chain N of Arg166.Moreover,these compounds also formed a conserved network of hydrophobic interactions with the surrounding residues,including Leu162,Asp164,Met208,Pro247,Pro248,and Tyr268.The molecular interaction analyses were found in agreement with the co-crystallized inhibitors of SARS-CoV-PLpro(PDB ID:3E9S and 3MJ5)[87](Table 1).

Fig.1.Multiple sequence alignments of SARS-CoV-2 PLprowith SRAS-CoV PLproand MERS-CoV PLpro.The conserved catalytic triad residues(Cys111-His272-Asp286)within SARSCoV-2,(Cys112-His273-Asp287)within SARS-CoV,and(Cys112-His275-Asp294)within MERS-CoV are highlighted with red color arrows.
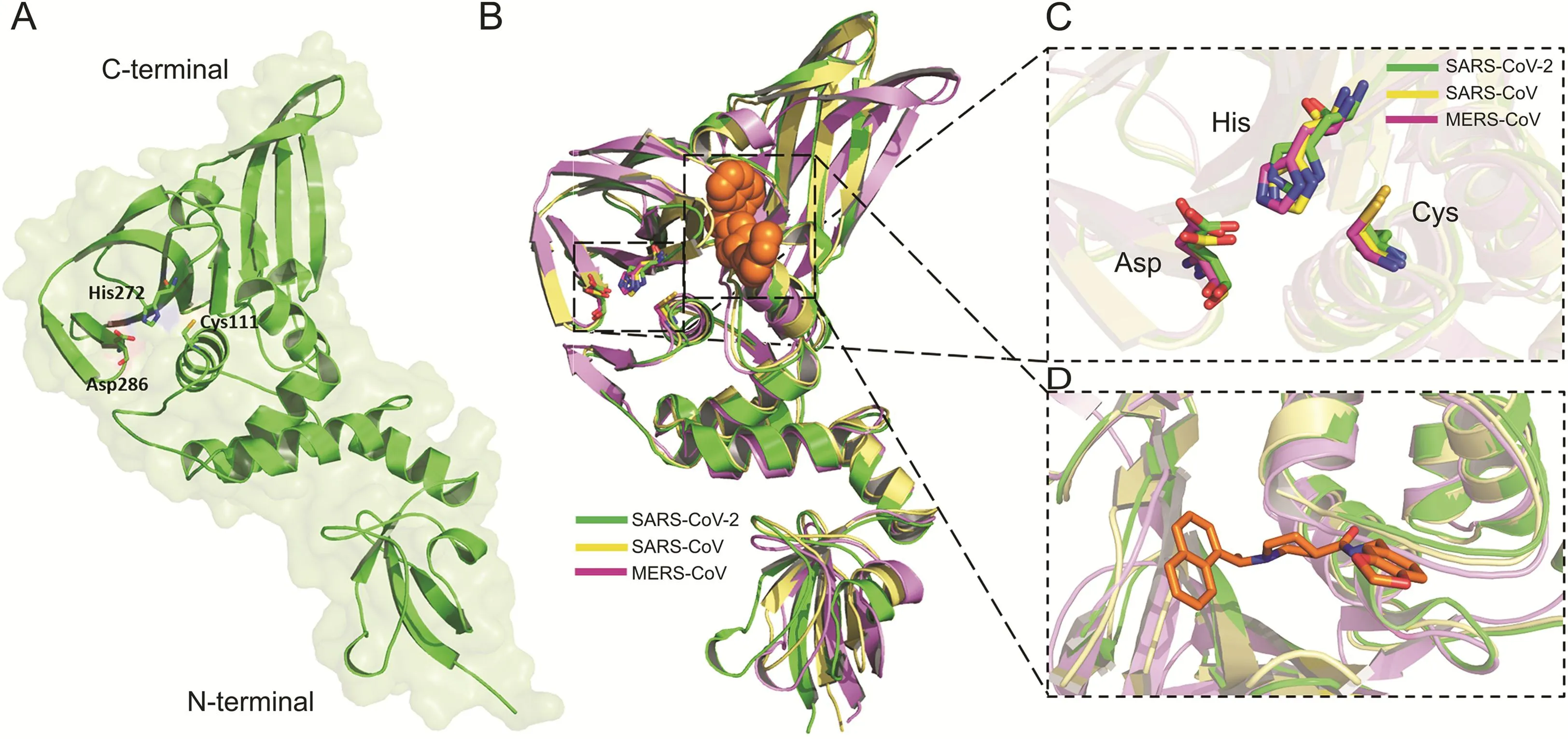
Fig.2.(A)The 3D structures of SARS-CoV-2 PLproenzymes.The catalytic triad residues Cys111-His272-Asp286 were shown green sticks.(B)The overlapping of the 3D structures of PLproenzymes of SARS-CoV-2(green)(PDB:6W9C),SRAR-CoV(yellow)(PDB:3MJ5)and MERS-CoV(purple)(PDB:4R3D).The conserved catalytic triad residues with each structure were shown in sticks.GRL-0667S within the binding site of SRAS-CoV PLprowas shown in orange spheres.(C)The overlapping of the catalytic triad residues of PLprowithin the active sites of SARS-CoV-2(green sticks),SRAR-CoV(cyan sticks),and MERS-CoV(pink sticks).(D)Close view to the targetable binding pocket within PLproenzyme based on the binding mode of GRL-0667S with SARS-CoV PLpro.
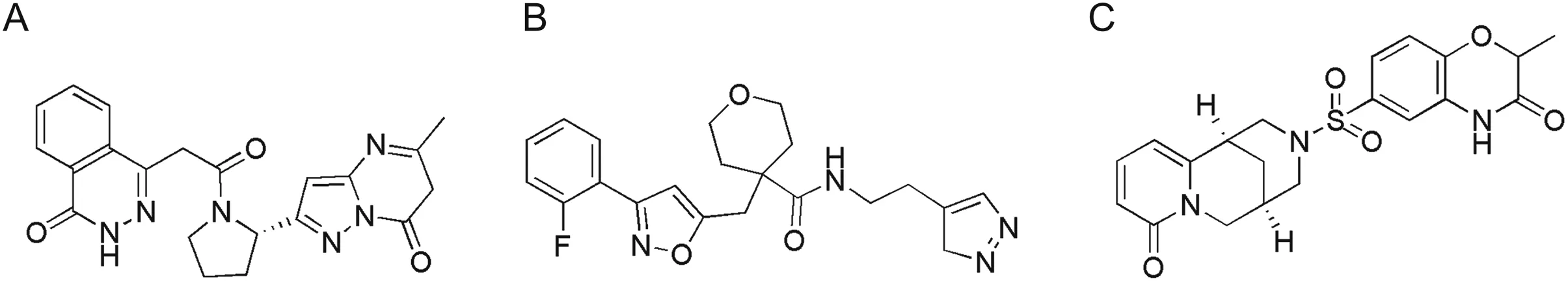
Fig.3.Chemical structures of screened hits;(A)ADM_13083841,(B)LMG_15521745 and(C)SYN_15517940.

Table 1Properties profile of candidate compounds.
3.4.ADMET screening and drug-ability results
ADMET-based drug scan tool at the SwissADME server was used to evaluate the drug-likeness of the proposed SARS-CoV-2 PLproinhibitors[74].ADM_13083841(C21H20N6O3)is a phthalazinone derivative with the log P value of 2.04 and molecular weight of 404.42 g/mol.The compound has six hydrogen-bond acceptor(HBA)and one hydrogen-bond donor(HBD)atoms.Another oxazole based compound selected from the docked molecules is LMG_15521745(C21H23FN4O3)with the log P value of 3.02 and molecular weight of 398.43 g/mol.It contains seven HBA atoms and one HBD.SYN_15517940(C20H21N3O5S)has a log P value of 2.30 and a molecular weight of 415.46 g/mol,together with six HBA atoms and one HBD(Table 2).
For further validation of the drug likeliness capability of target compounds,all these compounds were subjected to the PreADMET and pkCSM servers,which have five main parameters(absorption,distribution,metabolism,excretion,and toxicity)for assessment.These five parameters were then assessed according to the number of thresholds.All the three compounds successfully passed the criteria of drug-ability(Table 3).
3.5.MD simulation
MD simulation is a widely used computational method to analyze the dynamic behavior and stability of a ligand/protein complex under different conditions[30,53,88,89].All atoms MD simulations were carried out on the initial conformation of the hit compounds-PLprocomplexes obtained after the docking in the solvated states at 50 ns.
3.5.1.RMSD,RMSF and potential binding energy
The RMSD computes the direct changes in the atoms of superimposed proteins and is an acceptable approach to assess the stability of protein/ligand complexes[90-93].The RMSD values of the Cα-backbone atoms of SARS-CoV-2 PLproin complex to the three PLpropotential inhibitors were calculated with respect to the initial structure and compared with apo-protein in an unbound form.The RMSD values steadily increased in the beginning and remained converged throughout the simulation period,especially for ADM_13083841 and LMG_15521745 complexes.Similarly,the RMSD values of apo-protein reached an equilibrium after an initial increase within the first 5 ns and converged between 0.12 and 0.3 nm throughout the simulation course.The average RMSD values for the last 45 ns for apo-protein,ADM_13083841,LMG_15521745,and SYN_15517940 complexes were 0.18±0.03,0.18±0.03,0.19±0.04 and 0.18±0.03 nm,respectively(Fig.6A).
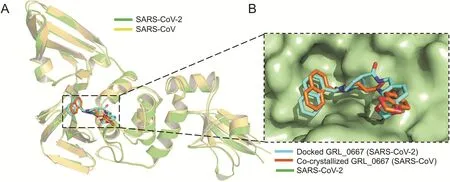
Fig.4.Validation of the docking protocol.(A)Ribbon representation for superimposition of docked GRL-0667S(cyan)into SARS-CoV-2 PLpro(green)(PDB:6W9C),and cocrystalized structure(orange)of GRL-0667S within the active site of SARS CoV PLpro(yellow)(PDB:3MJ5).(B)Superimposition of co-crystallized(orange)and best-docked pose(cyan)of inhibitor GRL-0667S in the active site of SARS-CoV-2 PLpro(PDB ID:6W9C,green molecular surface).
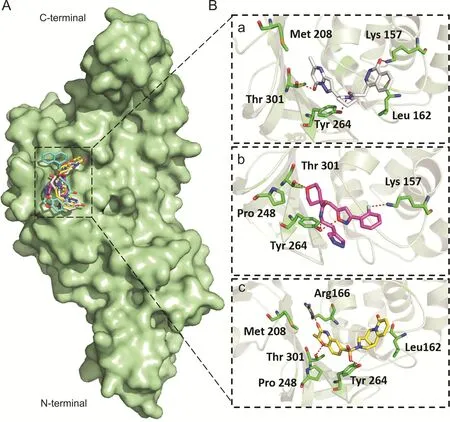
Fig.5.Binding modes and molecular interactions of screened compounds with SARS-CoV-2 PLpro(PDB:6W9C).(A)Surface representation of the binding mode of GRL-0667S(cyan),ADM_13083841(white),LMG_15521745(pink)and SYN_15517940(yellow)to SARS-CoV-2 PLpro(green).(B)The mechanism of molecular interactions of(a)ADM_13083841(white),(b)LMG_15521745(pink)and(c)SYN_15517940(yellow)to SARS-CoV-2 PLpro(green).
The RMSF measured the local protein flexibility and proved to be an excellent parameter to investigate the protein's residual flexibility over the simulation period[90,92].The time average of protein backbone RMSF values of the 315 amino acids of SARS-CoV-2 PLproprotein with and without the three candidate compounds was calculated over the 50 ns simulation period.Normal fluctuations in the constituent residues of SARS-CoV-2 PLprowere observed for apo-protein and all three complexes,PLpro-ADM_13083841,PLpro-LMG_15521745 and PLpro-SYN_15517940,and were plotted to compare the residual flexibility.As shown in Fig.6B,major fluctuations were observed mainly in the loop regions(residue nos.185-200 and 220-230),located away from the binding pocket.The average RMSF values were calculated as 0.09±0.05,0.11±0.05,0.11±0.06 and 0.09±0.04 nm for apo-protein,ADM_13083841,LMG_15521745and SYN_15517940,respectively.
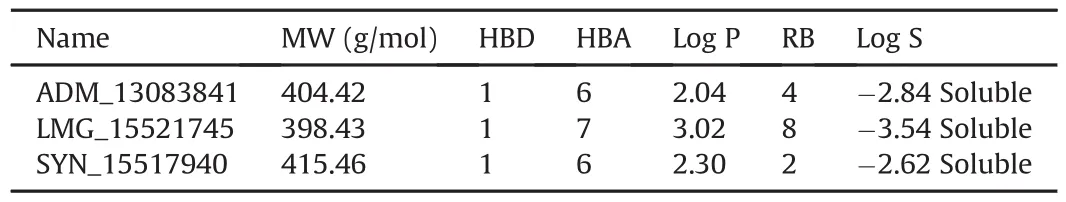
Table 2Drug-likeness properties of identified compounds.
Furthermore,the potential energy over the simulation time for the three complexes and apo-protein was calculated.The results indicated that the three complexes remained in a stable pattern throughout the 50 ns simulations,as shown in Fig.6C.These RMSD,RMSF,and potential energy MD simulation results confirmed the stability of all selected compounds at the catalytic-site of SARSCoV-2 PLpro.
3.5.2.Rg,hydrogen bond interaction and SASA analysis
The Rg is a parameter for evaluating the behavior and stability of the biological systems during the MD trajectories by measuring the compactness of biomacromolecules' structures[94,95].The Rg can also be used as an indicator of whether the complex will maintain folded conformation after the MD simulation.The Rg values of the three complexes and apo-protein were stabilized after the initial increase at 5 ns,supporting the RMSD results that systems had reached the equilibrium state(Fig.7A).The average Rg values for the three complexes and apo-protein remained relatively consistent for the last 45 ns,indicating a stably folded structure with an average value of 2.32±0.01,2.32±0.02,2.32±0.02 and 2.31±0.01 nm for apo-protein,ADM_13083841,LMG_15521745,and SYN_15517940,respectively.
Hydrogen bonds play a crucial role as stabilizing forces for a ligand-protein complex[90-92].The total number of hydrogen bonds formed between the three candidate compounds and SARSCoV-2 PLprois shown in Fig.7B.All candidate compounds can makeup to four stable hydrogen bonds with SARS-CoV-2PLprothroughout the simulation period supporting the docking results.These bonding parameters represented that all candidate compounds may bind effectively and tightly to the SARS-CoV-2 PLpro.
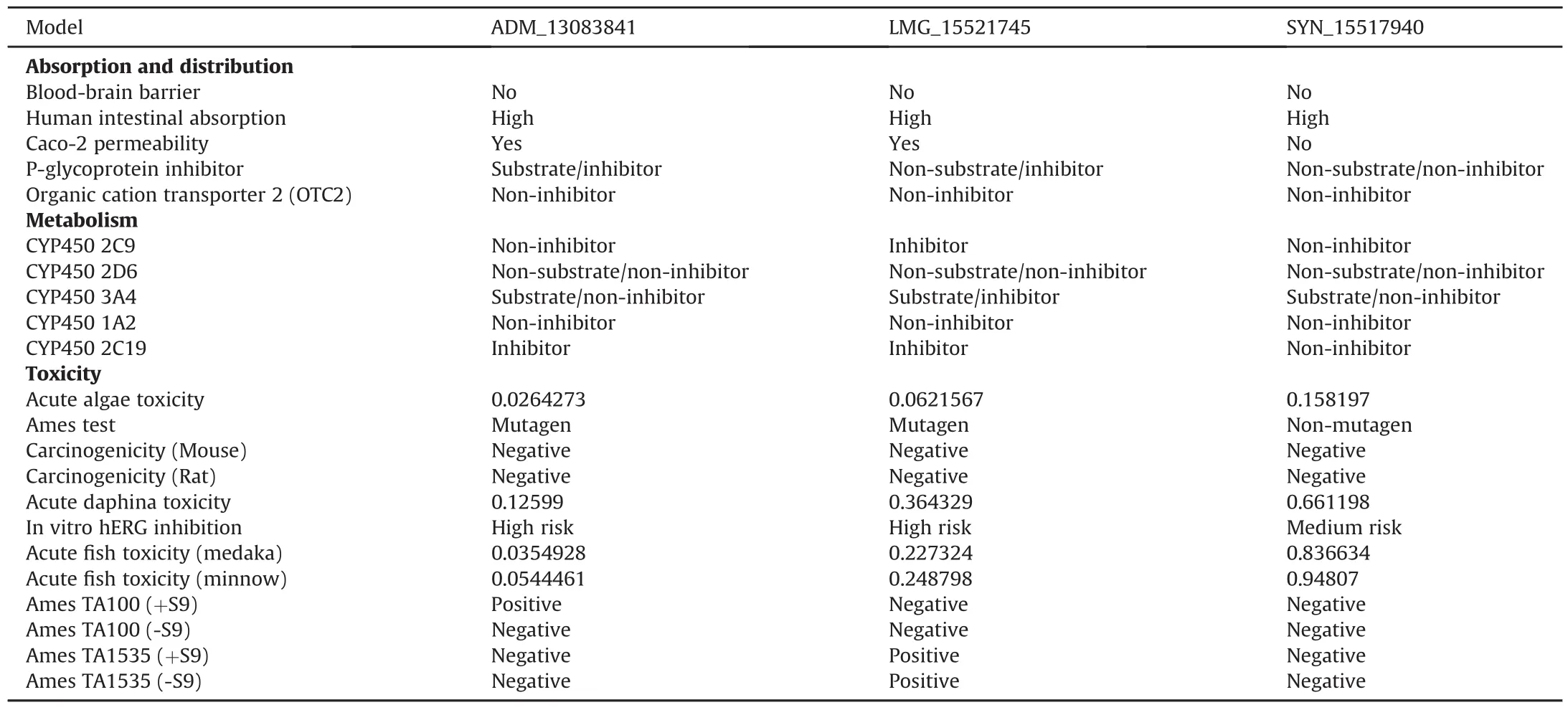
Table 3ADMET profiling for absorption,metabolism and toxicity parameters of identified compounds.
The SASA is a tool to measure the water accessible proportion of the biomolecule surface[96].The calculation of SASA value is a useful tool to predict the degree of the conformational changes which resulted due to complex interaction.The calculated average SASA values for the last 45 ns simulation time were 165.27±1.22,164.78±1.21,165.18±1.18,and 165.51±1.27 nm2,respectively(Fig.7C).These results suggested there were no observed changes in the accessibility area of all three systems over the 50 ns simulation time.Thus,in terms of SASA analysis,the relative stability of our protein-ligand complexes has been concluded.
3.5.3.PCA
The PCA analysis was performed to identify conformational changes that accompanied the ligands binding within different protein/ligand complexes.In the current study,the first two principal components(PC1 and PC2)were selected for predicting the protein motions.The projection of two eigenvectors for apo-protein as well as all three complexes,PLpro-ADM_13083841,PLpro-LMG_15521745,and PLpro-SYN_15517940 is shown(Fig.8).Generally,the complex that occupies a more phase-space with a nonstable cluster and complex that occupies less phase-space with a stable cluster represented a less stable complex and a more stable complex,respectively.From the graphs,the apo-protein,as well as the three complexes,occupied less phase-space.The clusters shifting were from-4 to 5 nm in case of apo-protein,-8 to 6 nm in case of ADM_13083841.In contrast,the clusters shifting were from-6 to 6 and-4 to 4 in cases of LMG_15521745 and SYN_15517940,respectively.With respect to apo-protein values,all complexes showed a very stable complex.
3.6.Predicted binding free energy calculations
The MM/GBSA and MM/PBSA are both end-point methods and present more physically meaningful information than docking scoring functions.These methods have been extensively utilized in the identification of potential antiviral inhibitors[83,84,97,98].The absolute energy of binding(ΔGbind)of all three hit compounds together with co-crystallized PLproinhibitor,GRL-0667S[87],was predicted through mechanics/Poisson-Boltzmann(generalized born)surface area(MM/PB(GB)SA)method on 50 snapshots extracted from the last 20 ns of the simulation period.It is important to note that we also incorporated entropic contributions(-TΔS)in ligand binding,which are computationally expensive in the MMPBSA method and give better accuracy[99].Moreover,entropy effects play an essential role in protein-ligand interactions[100].The calculations are tabulated in Table 4.
According to the MM/PB(GB)SA calculations,van der Waals(ΔEvdW)interaction was the main driving force in complex stabilization with ADM_13083841 (ΔEvdW= -46.12 kcal/mol),LMG_15521745(ΔEvdW=-41.3 kcal/mol)and SYN_15517940(ΔEvdW=-39.88 kcal/mol),and was about 1-2 fold stronger than electrostatic attraction energy(ΔEele)predicted to be-14.1,-16.24,and 15.3 kcal/mol,respectively.Therefore,it was found that compounds interacted mainly through van der Waals interactions and these findings are in agreement with the co-crystalized GRL-0667S(ΔEvdW=-43.7 and ΔEMM=-12.39)[87].Together with the solvation effect in ADM_13083841 (ΔGsol(PBSA)= 30.83;ΔGsol(GBSA)=24.03 kcal/mol),LMG_15521745(ΔGsol(PBSA)=32.15;ΔGsol(GBSA)=25.48kcal/mol)and SYN_15517940(ΔGsol(PBSA)=26.17;ΔGsol(GBSA)=20.32 kcal/mol)complexed with SARS-COV-2 PLproand incorporation of entropic terms,the absolute ΔGbindaccounted for-5.09(ADM_13083841),-4.17(LMG_15521745)and-7.11 kcal/mol(SYN_15517940)as per MM-PBSA(ΔGbind(MM/PBSA))approach and values of 11.89(ADM_13083841),-10.84(LMG_15521745)and-12.96 kcal/mol(SYN_15517940)were taken from the MMPBSA(ΔGbind(MM/PBSA))approach.The obtained ΔGbindvalues of all three compounds were in agreement with the co-crystalized GRL-0667S,which revealed relatively similar values by MM/PBSA(-5.81 kcal/mol)and MM/GBSA(-9.82 kcal/mol)methods.
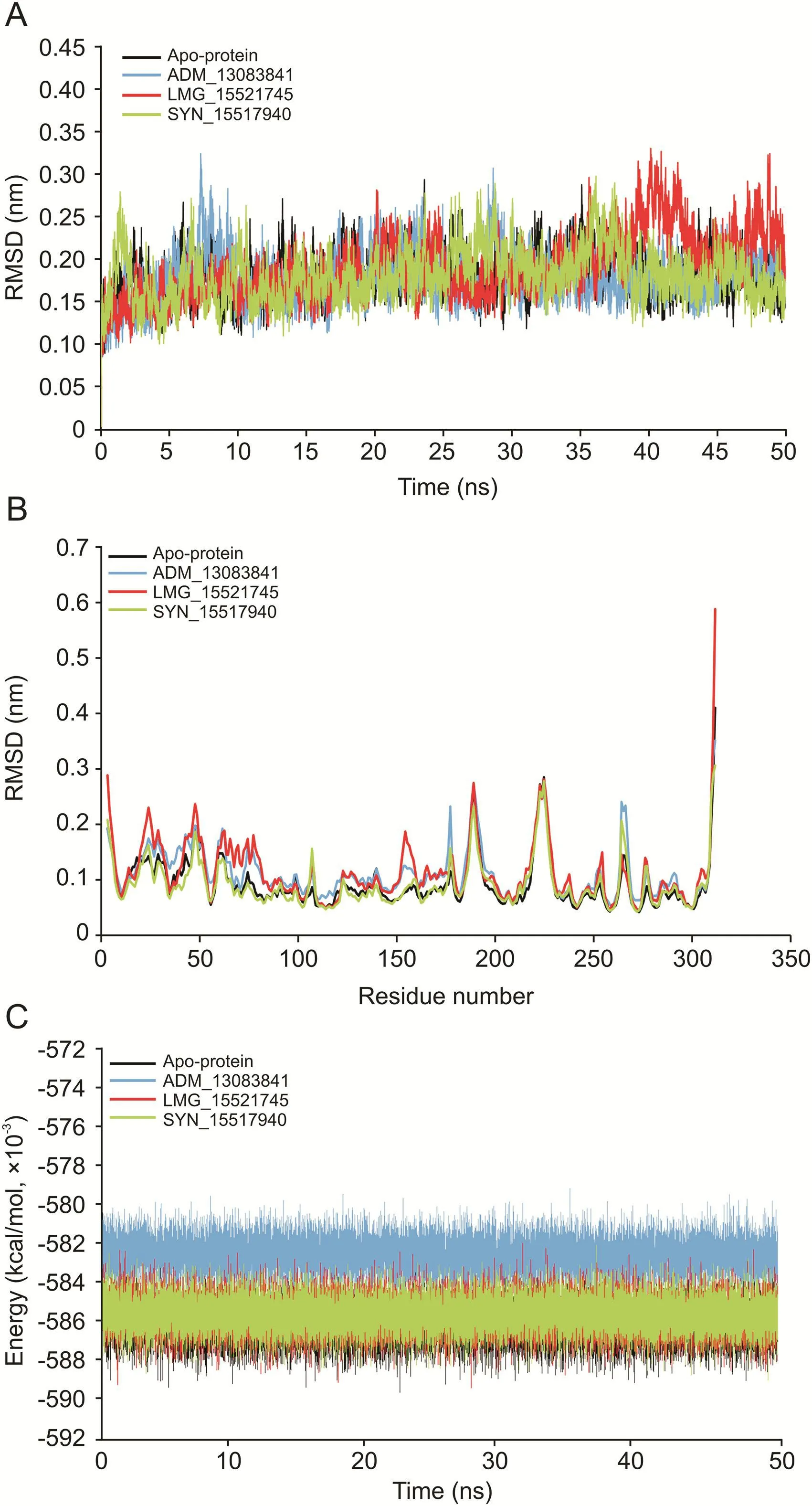
Fig.6.(A)The root mean square deviation(RMSD)of C-Cα-N backbone vs.simulation time for solvated SARS-CoV-2 PLproin apo-state and in complex with the three candidate compounds over the time of 50 ns MD simulation.(B)The root mean square fluctuation(RMSF)of SARS-CoV-2 PLproin apo-state and in complex with the three candidate compounds.(C)The potential energy for SARS-CoV-2 PLproin apo-state and in complex with the candidate compounds over the time of 50 ns MD simulation.
3.6.1.Per-residue decomposition analysis
In order to evaluate the binding role of key interacting residues of the active site,(ΔGresidue)calculations were performed using the MMGBSA method.The total energy decomposition values associated with ligand binding is presented in Fig.9.We highlight here only the significant energy contributing residues in ligand binding.Briefly,the obtained results revealed that residues,including Asp164,Met208,Pro247,Pro248,Tyr264 and Thr301 located in the active site of SARS-COV-2 PLpro,were found important for interactions with ADM_13083841, LMG_15521745, and SYN_15517940.Among these,the residual decomposition analysis revealed the favorable contributions(<-1.7 kcal/mol)of Asp164,which exhibited ΔGresidueof-1.92,-1.86,and-1.74 kcal/mol,Tyr264 which exhibited ΔGresidueof-2.14,-2.08,and-2.81 kcal/mol with ADM_13083841,LMG_15521745 and SYN_15517940,respectively.Moreover,Tyr264 was also found interacting through H-bonds in all three complexes(Fig.5).
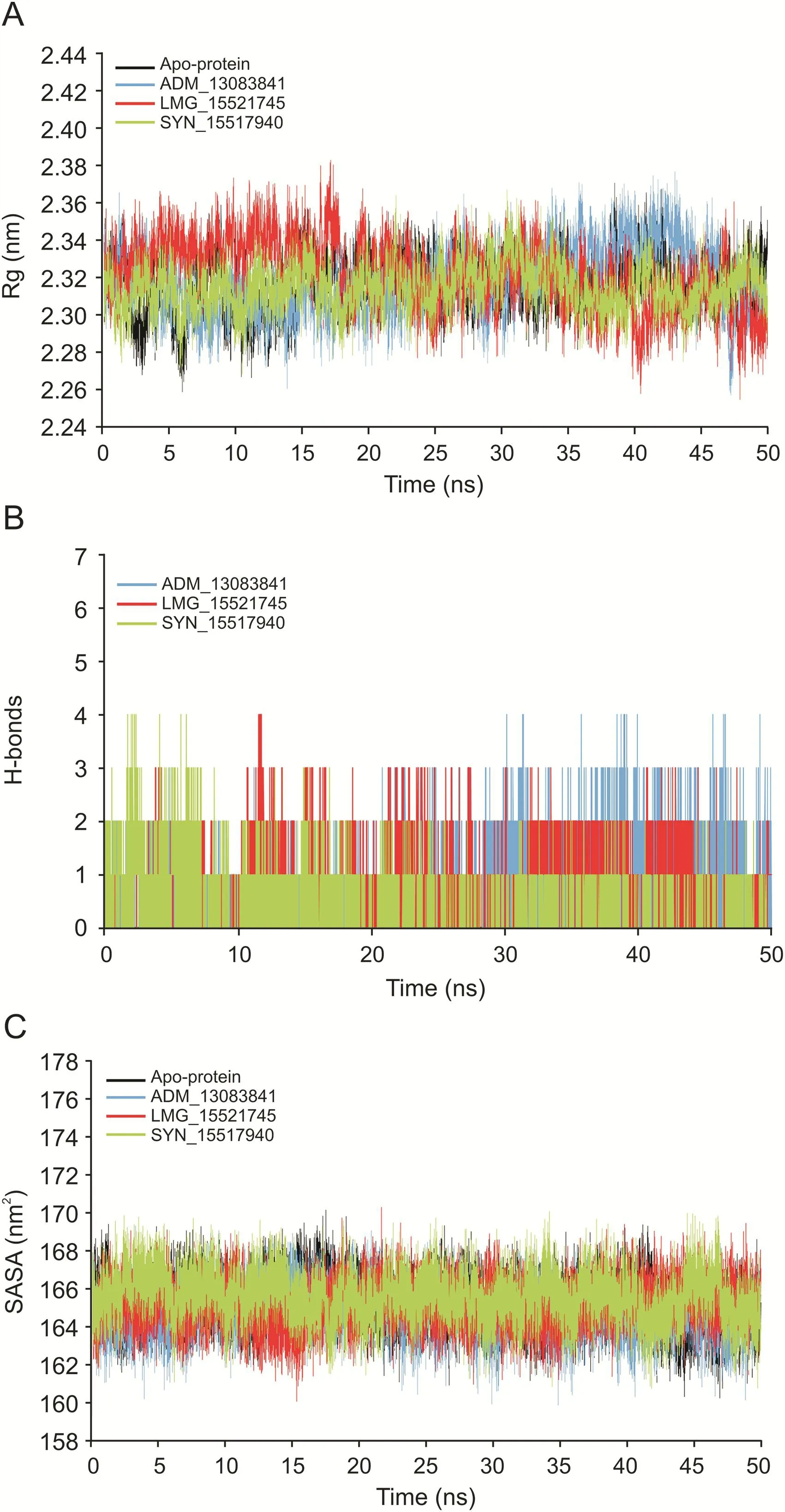
Fig.7.(A)The radius of gyration(Rg)of SARS-CoV-2 PLproin apo-state and in complex with the three candidate compounds during 50 ns MD simulation.(B)Plot of number of hydrogen bond within the SARS-CoV-2 PLproin complex with the three candidate compounds during 50 ns MD simulation.(C)Plot of solvent accessible surface area(SASA)of SARS-CoV-2 PLproin apo-state and in complex with the three candidate compounds over the time of 50 ns MD simulation.
In comparison,the ΔGresiduevalues of co-crystallized GRL-0667S showed a relatively similar trend.Hence,ΔGresidueenergies of highly interacting residues suggested that the molecular structures of all three compounds fitted well inside the active-site of SARSCOV-2 PLpro.Moreover,the obtained results after residual decomposition analysis were in accordance with the complex stability analysis through MD simulation,where the stable RMSD was achieved due to these pairwise interactions throughout the simulation period.Hence,we hypothesized that these critically important active site residues might be crucial in inhibitor recognition for the development of SARS-COV-2 PLproinhibitors and could lead to further optimization of these compounds.
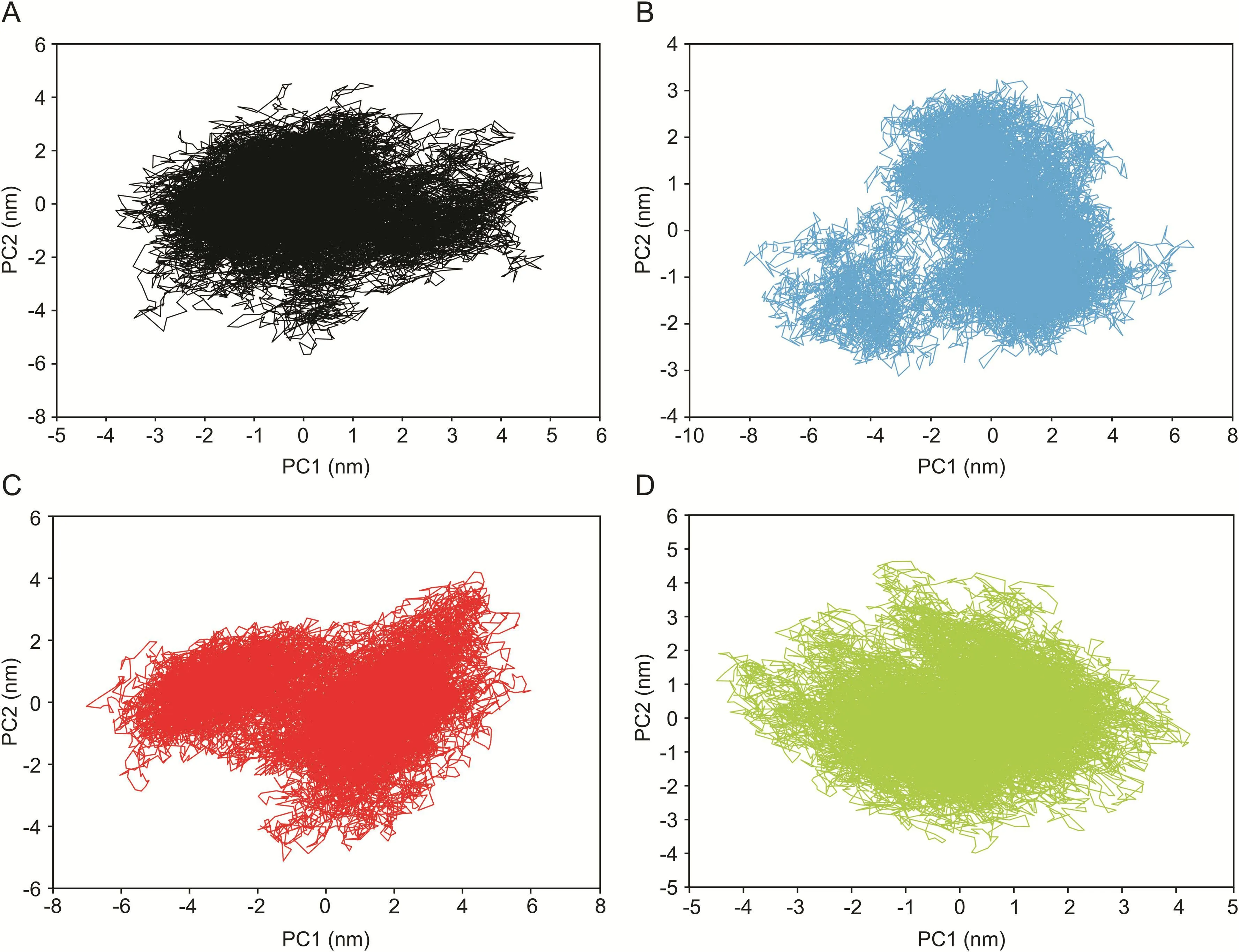
Fig.8.Two-dimensional principle component analysis(PCA)projections of trajectories obtained from 50 ns MD simulations.(A)Apo-protein,(B)ADM_13083841,(C)LMG_15521745,and(D)SYN_15517940.
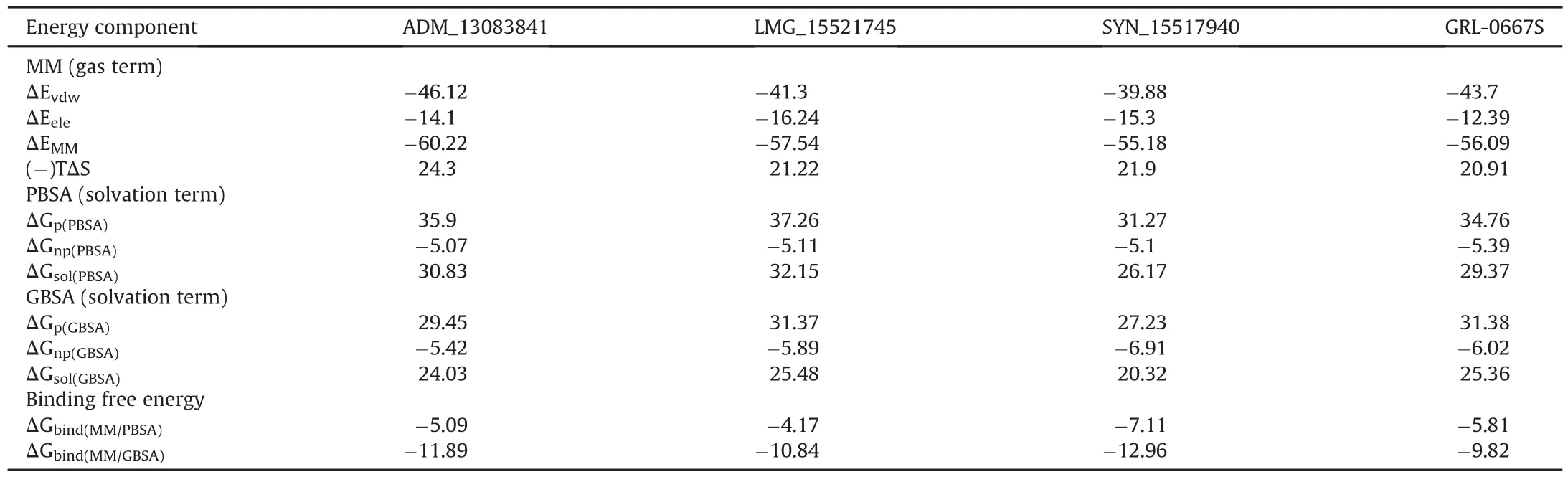
Table 4ΔGbindvalues of ADM_13083841,LMG_15521745 and SYN_15517940 in complex with SARS-CoV-2 PLprocalculated by the MM/PB(GB)SA method.
3.7.Molecular docking of screened hits with SARS-CoV and MERSCoV PLpros
Disulfiram is an FDA approved drug used to treat chronic alcoholism.Previous research reported that disulfiram can inhibit SARS-CoV and MERS-CoV PLprosas an allosteric,competitive or mixed inhibitor[101].Disulfiram has also been proposed for the treatment of SARS-CoV-2[102-104].Similarly,previous studies reported that lopinavir showed promising results against MERSCoV[105-108]and SARS-CoV[109-111],and currently it is under clinical trials for COVID-19 treatment[11,112-114].Lopinavir is an FDA approved serine protease inhibitor used to treat HIV-1 infection[115].Therefore,to test the hypothesis and validate the possibility of pan-PLprobased broad-spectrum inhibitors,next we determined the ability of the selected compounds to bind to the SARS-CoV and MERS-CoV PLpros,by the validated flexible docking approach into the same target site.Interestingly,these compounds showed high binding affinity toward SARS-CoV PLprowith binding energy scores ranged from-8.7 to-7.9 kcal/mol(Fig.10).However,the binding energy scores of these compounds were low in the case of MERS-CoV,ranging from-5.9 to-6.7 kcal/mol,indicating that their activity toward SARS-CoV might be greater than the activity against MERS-CoV(Fig.11).This could be due to the structural difference of MERS-CoV blocking loop 2[116].
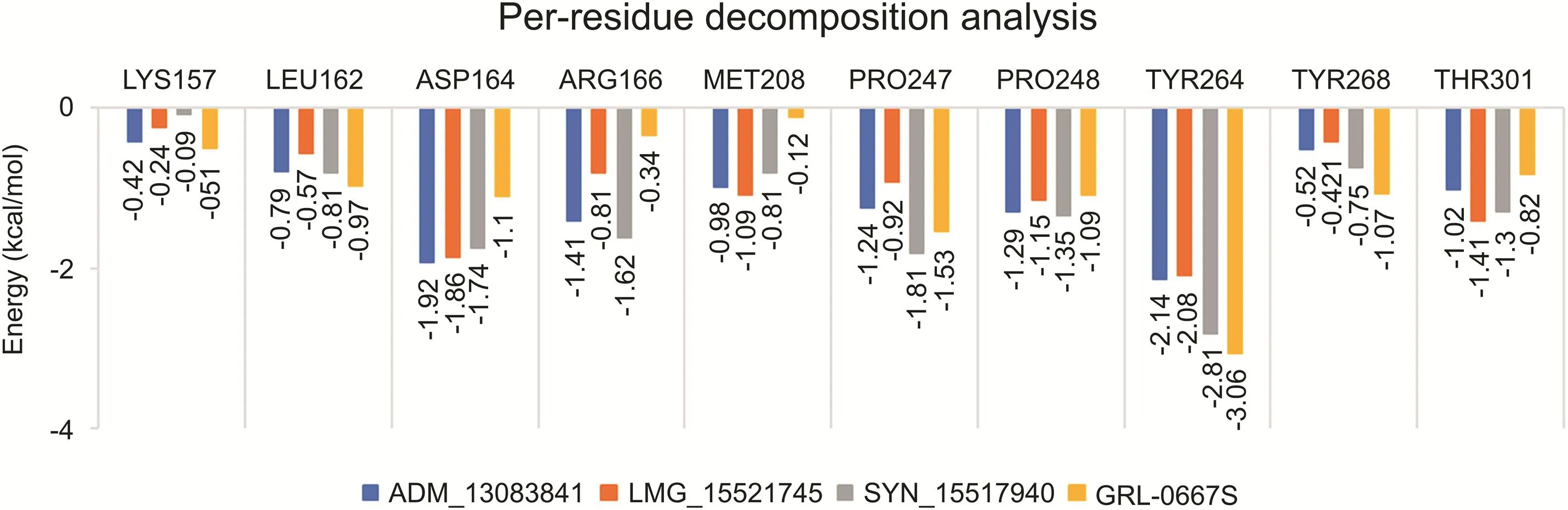
Fig.9.Per-residue decomposition analysis using MM-GBSA methods and highly interacting binding site residues are displayed together with ΔGresiduederived from last 20 ns of MD simulations.
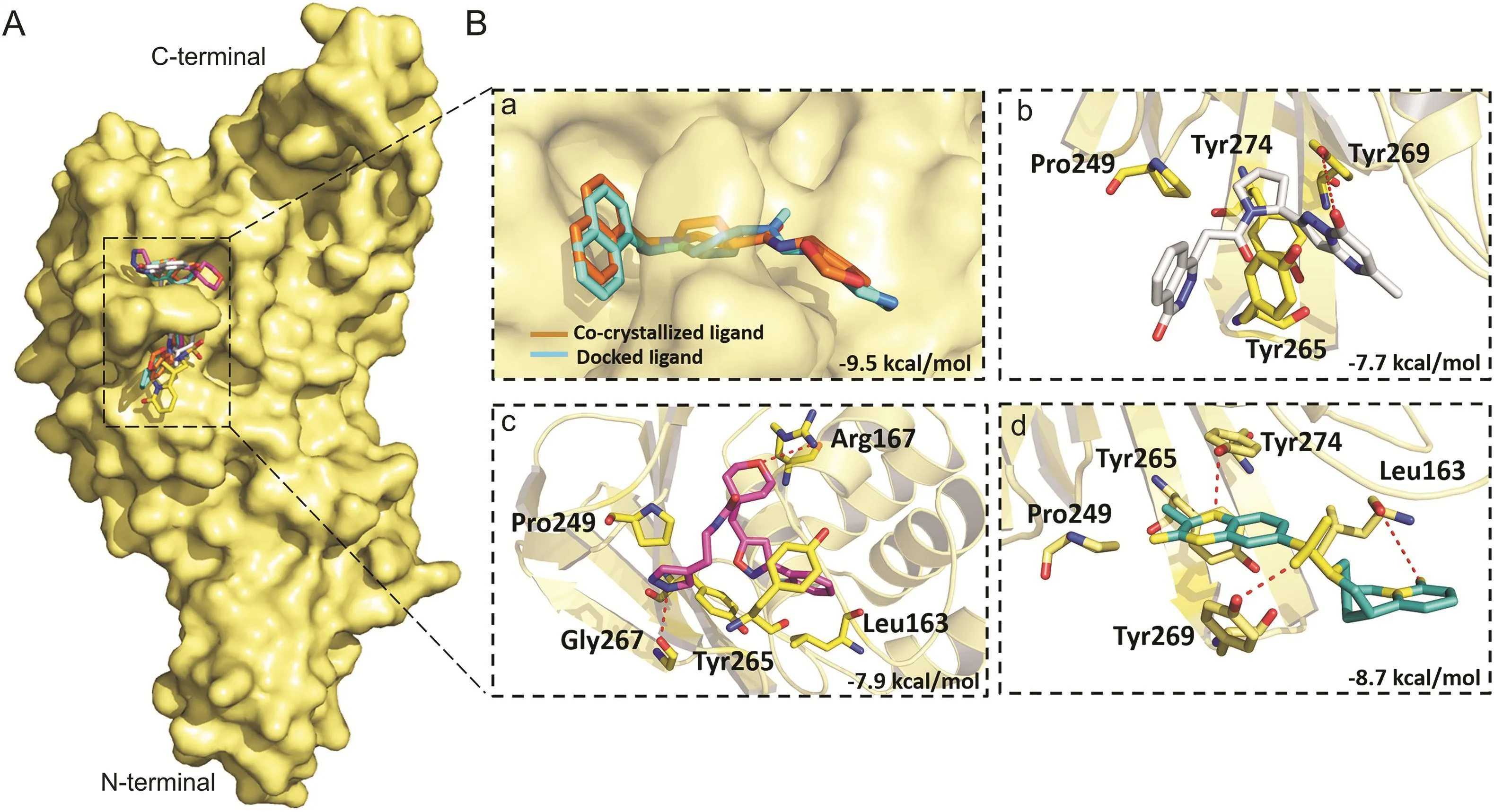
Fig.10.Binding modes and molecular interactions of screened compounds with SARS-CoV PLpro(PDB:3MJ5).(A)Surface representation of the binding mode of co-crystalized GRL-0667S(cyan),docked GRL-0667S(orange),ADM_13083841(white),LMG_15521745(pink)and SYN_15517940(green)to SARS-CoV PLpro(yellow).(B)The mechanism of molecular interactions of compounds to SARS-CoV PLpro.Close view to the binding mode of(a)co-crystalized GRL-0667S(cyan)and docked GRL-0667S(orange)to SARS-CoV PLpro,(b)ADM_13083841(white),(c)LMG_15521745(pink)and(d)SYN_15517940(green)to SARS-CoV-2 PLpro.
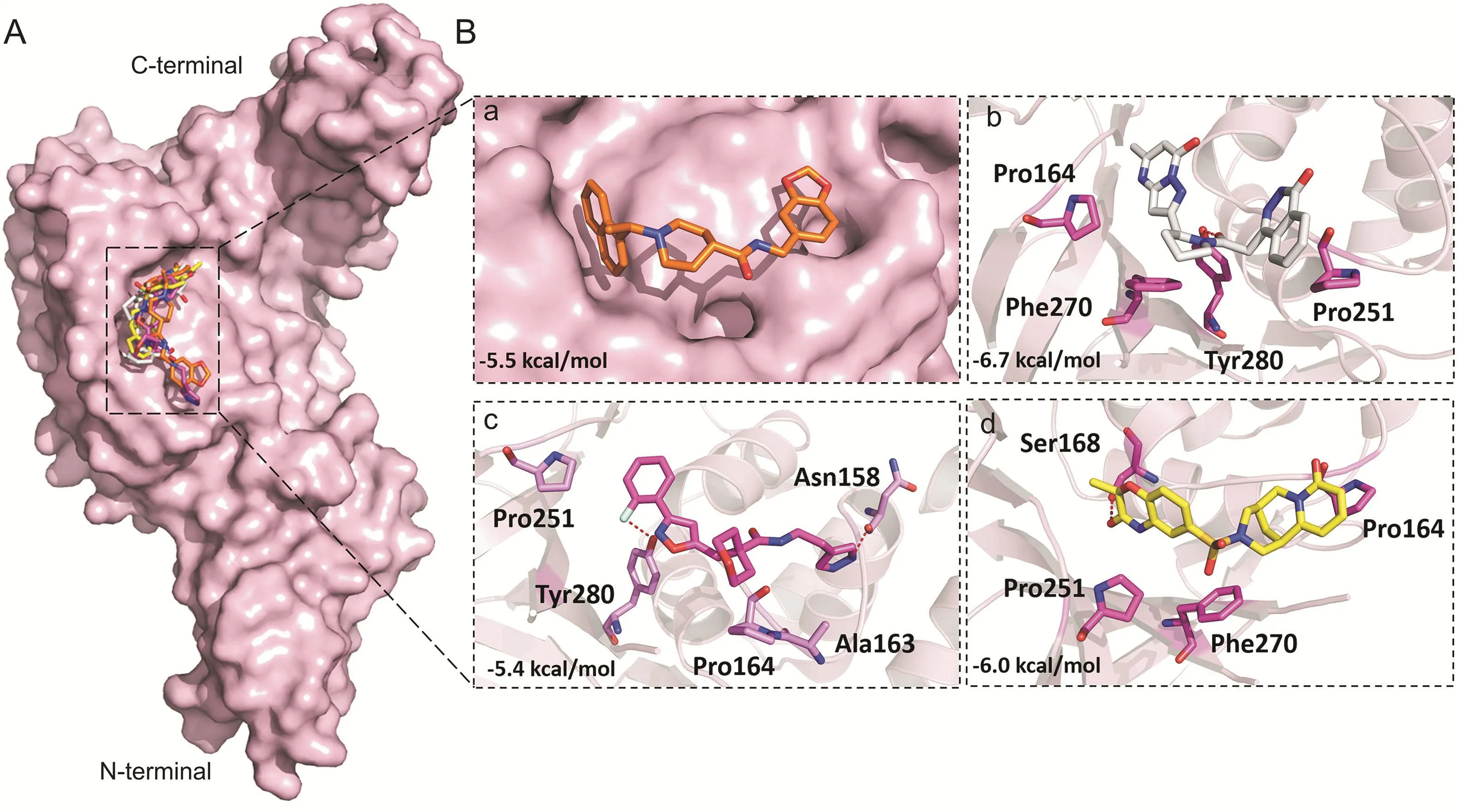
Fig.11.Binding modes and molecular interactions of screened compounds with MERS-CoV PLpro(PDB:4R3D).(A)Surface representation of the binding mode of docked GRL-0667S(orange),ADM_13083841(white),LMG_15521745(pink)and SYN_15517940(yellow)to MERS-CoV PLpro(pink).(B)The mechanism of molecular interactions of compounds to MERS-CoV PLpro.Close view to the binding mode of(a)docked GRL-0667S(orange),(b)ADM_13083841(white),(c)LMG_15521745(pink)and(d)SYN_15517940(green)to MERSCoV PLpro.
Our effort to target the deadliest human CoVs(SARS-CoV,SARSCoV-2 and MERS-CoV)PLproconcurrently by investigating their conservation(structural and functional)produced significant results.The present study identified three small-molecule protease inhibitors with great capability of drug leads,capable of inhibiting PLproof SARS-CoV-2.These set of compounds could function as broad-spectrum pan-PLproinhibitors against deadly human CoVs infections.This is critically important against constantly evolving CoVs.The benefits of treatment strategies involving pan-inhibitors have already been reported in the case of Dengue virus[28,117],HCV[118-121],and HIV[122].The screened inhibitors in the present study may lead towards a medicinal solution against the variety of constantly evolving CoVs by effectively targeting/hindering the proteolytic role of their PLpros.Therefore,our findings warrant further experimental work on screened pan-PLproinhibitors for further drug optimization.
4.Conclusions
This comprehensive study offers an integrated computational approach towards the discovery of three novel hit compounds,screened from a focused library of~7,000 compounds having a diverse scaffold that can specifically target SARS-COV-2 PLpro,which permits in vitro evaluations.These three compounds,ADM_13083841,LMG_15521745,and SYN_15517940,were selected for further computational studies to gain a deep insight into their binding mode,mechanism of molecular interaction,and ADMET analysis.The in-depth structural exploitation of key residues of the active site together with the dynamic scaffolds of hit compounds adopted after 50 ns MD simulations offered the way to design broad-spectrum inhibitors against deadly human CoVs.The structural/functional conservation of SARS-CoV,MERS-CoV,and SARSCoV-2 revealed strikingly similar conformations of active site residues.Altogether,the identification of highly contributing residues towards overall ligand binding energy might provide an excellent platform to further enhance the inhibitor recognition potential of SARS-COV-2 PLpro.Although the present study lacks in silico binding mode validation,the structural evidence obtained from this computational study has surfaced the way in the designing of pan-PLprobased inhibitors as broad-spectrum antiviral agents to combat SARS-CoV-2 and other pathogenic human coronaviruses.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the Starting Research Grant for High-level Talents from Guangxi University and the Postdoctoral Project from Guangxi University.Authors would like to thank Guangxi University,Prince Sattam Bin Abdulaziz University and University of Leuven for providing necessary facilities to conduct this research.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.08.012.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Challenges for cysteamine stabilization,quantification,and biological effects improvement
- Offline two-dimensional liquid chromatography coupled with ion mobility-quadrupole time-of-flight mass spectrometry enabling fourdimensional separation and characterization of the multicomponents from white ginseng and red ginseng
- Single-run reversed-phase HPLC method for determining sertraline content,enantiomeric purity,and related substances in drug substance and finished product
- Development of a UHPLC-MS/MS method for the quantification of ilaprazole enantiomers in rat plasma and its pharmacokinetic application
- Use of subcutaneous tocilizumab to prepare intravenous solutions for COVID-19 emergency shortage:Comparative analytical study of physicochemical quality attributes
- Vancomycin pretreatment attenuates acetaminophen-induced liver injury through 2-hydroxybutyric acid