Single-run reversed-phase HPLC method for determining sertraline content,enantiomeric purity,and related substances in drug substance and finished product
2021-01-21AlessiRosettiRosellFerrettiLeoZnittiAdrinoCsulliCludioVillniRoertoCirilli
Alessi Rosetti,Rosell Ferretti,Leo Znitti,Adrino Csulli,Cludio Villni,Roerto Cirilli,*
aNational Centre for the Control and Evaluation of Medicines,Chemical Medicines Unit,Istituto Superiore di Sanità(ISS),Viale Regina Elena 299,00161,Rome,Italy
bDepartment of Chemistry and Technology of Drugs,Sapienza University of Rome,00185,Rome,Italy
cWHO Collaborating Centre for the Epidemiology,Detection and Control of Cystic and Alveolar Echinococcosis,European Union Reference Laboratory for Parasites,Istituto Superiore di Sanità(ISS),Viale Regina Elena 299,00161,Rome,Italy
ABSTRACT
Keywords:
Chiralpak IG-3
Enantiomers
Diastereomers
Reversed-phase enantioselective HPLC
Sertraline HCl
1.Introduction
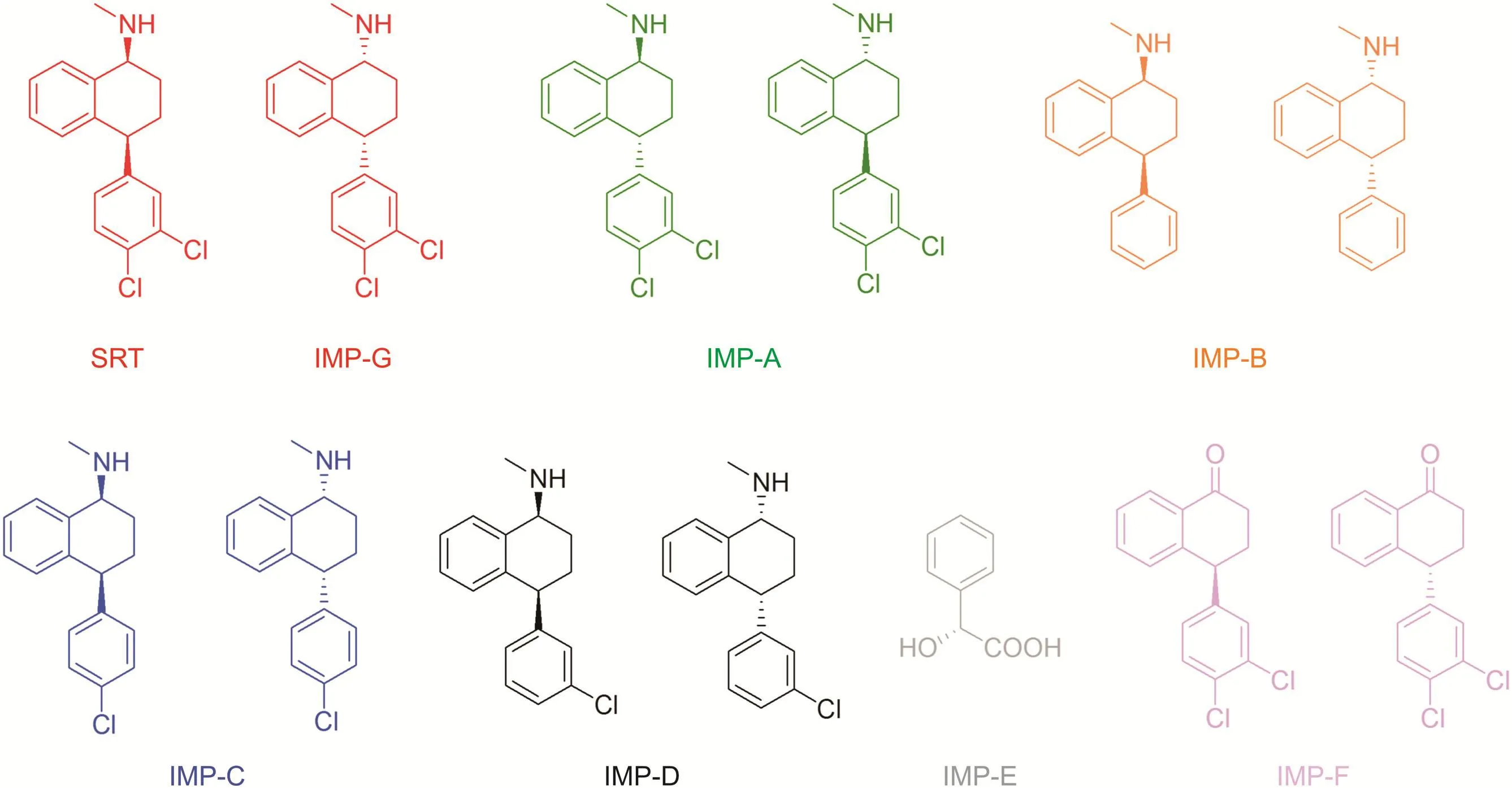
Fig.1.Chemical structures and abbreviations of sertraline and its potential organic impurities.
High-performance liquid-chromatography(HPLC)on a chiral stationary phase(CSP)is widely recognized as an important analytical tool for determining the enantiomeric purity of chiral drugs[1].The popularity of enantioselective HPLC essentially depends on the availability of commercial CSPs capable of resolving a wide range of chiral compounds under multimodal elution conditions,without preliminary chiral derivatization.In addition,this method provides the desired sensitivity for trace-level determinations(i.e.,the typical detection limits for organic impurities are 0.1% or lower)and a high flexibility in the choice of the detector(e.g.,UV,MS,and chiroptical detectors).As reported by drug regulatory agencies,the development and production of enantiopure drugs require:i)marked differences in at least one of pharmacodynamic,pharmacokinetic,and toxicological properties of drug enantiomers;ii)appropriate manufacturing control of synthesis and impurities;and iii)efficient routes for evaluating chiral and achiral related substances in either drug substance or drug product[2].Among the enantiopure chiral drugs currently in use,sertraline HCl(SRT)is one of the most successful and marketed drugs in the world.It has been estimated that over 41 million of SRT prescriptions were issued in 2013 only in the United States[3].SRT is the prototype of drugs belonging to the group of selective serotonin reuptake inhibitors(SSRIs)in the brain.It displays two stereogenic centers,butonly the stereoisomer with (1S,4S)configuration is clinically effective for the treatment of depression as well as panic,social anxiety,and obsessive-compulsive disorders[4].The cis-(1R,4R)enantiomer of SRT(IMP-G)and the diastereomeric pair of trans-enantiomers(IMP-A)are therefore considered as impurities.The chemical structures of the stereoisomeric impurities of SRT are shown in Fig.1.According to the monograph reported in the European Pharmacopoeia(EP)[5],the enantiomeric excess(ee)of SRT in the active pharmaceutical ingredient(API)is evaluated by an enantioselective HPLC method based on a polysaccharide-based Chiralpak AD-H(250 mm ×4.6 mm,5μm)column with a mobile phase containing n-hexane,2-propanol,and diethylamine(DEA).Although the current official method enables the baseline enantioseparation of SRT in normal-phase conditions,the quantitative analysis of the enantiomeric purity of SRT in the presence of its related chiral substances has yet to be fully demonstrated.In the same monograph,the SRT and(R)-mandelic acid(IMP-E)levels are evaluated using two chromatographic methods based on different stationary phases,whereas the content of other impurities is assessed by gas chromatography.Thus,the strict structural similarity between the parent drug and related substances makes the SRT analysis time consuming and expensive.A convenient approach to reduce the solvent consumption and simplify the analytical procedure is to use enantio-,diastereo-,and chemo-selective conditions within reasonably short retention times.“Single-run”approach is especially challenging and requires highly selective and efficient chiral stationary phases.Rao et al.have successfully applied this strategy for the determination of the enantiomeric purity of SRT in the presence of diastereomeric impurities,as well as of three other potential chiral impurities(IMP-B,IMP-C,and IMP-D,Fig.1),in drug substance and finished product.The reversed-phase method was developed and validated using the CYCLOBOND I 2000 DM column(250 mm×4.6 mm,5μm)packed with a heptakis(dimethyl-β-cyclodextrin)-based stationary phase[6].The CYCLOBOND I 2000 DM columnwas also used to determine sertraline enantiomers in rat plasma using a two-dimensional liquid chromatography/electrospray ionization-tandem mass spectrometry system[7].The aim of this work was to develop an alternative single-run stereoselective HPLC method capable of determining related chiral substances in SRT samples with a limit of quantification of 0.1% or below.We investigated all impurities listed in the EP monograph(Fig.1),with the only exception of IMP-D;the immobilized polysaccharide-based Chiralpak IG-3 CSP was used as packing material.In the Chiralpak IG-3 CSP,the amylose tris(3-chloro-5-methylphenylcarbamate)(ACMPC)polymeric selector was immobilized onto 3-μm silica particles[8].Recent studies showed that meta-substituted ACMPC-based CSPs can be successfully applied to develop enantioselective HPLC protocols useful for the resolution of biologically active chiral compounds[9-11]in both normal-phase and aqueous modes[12,13].In addition,the ACMPC selector was also shown to be effective for the simultaneous separation of structurally related chiral analytes[8,12-16].This ability can be particularly useful in ensuring both enantio-and chemo-selective conditions for the assay and determination of related substances in APIs.Since for the HPLC analysis of compounds of pharmaceutical interest,the use of a mobile phase containing water presents clear advantages over normal-phase modes(e.g.,better solubility of drugs in the mobile phase,compatibility with the LC-MS configuration)[14,17],the chiral discrimination ability of the Chiralpak IG-3 CSP was optimized using methanol-water-DEA and acetonitrile-water-DEA mixtures as mobile phases.Finally,the developed stereoselective protocol was applied to the determination of the content of SRT and its related substances in commercially available tablets.
2.Experimental
2.1.Reagents and chemicals
SRT and SRT for peak identification(SRT-Peak-ID)EP standards were purchased from the European Directorate for the Quality of Medicines and Healthcare(EDQM)(France).The impurities A((1R,4S),HCl salt,potency 90.7%),C(unknown absolute configuration,HCl salt,potency 98.4%),E((R),potency 100%),F(racemic mixture,HCl salt,potency 98.5%),and G((1R,4R),HCl salt,potency 98.6%)(Fig.1)and placebo were provided by Mylan Pharmaceutical Company(Canonsburg,Pennsylvania,US).HPLC-grade solvents were used as supplied by Aldrich(Milan,Italy).HPLC analyses were carried out on a Chiralpak IG-3(250 mm×4.6 mm,3μm)column(Chiral Technologies Europe,Illkirch-Graffenstaden,France).SRT oral tablets were purchased from a local drugstore.
2.2.Instruments
The HPLC system consisted of a Waters Alliance e2695 separation module and 2998 photodiode array detector(PDA).Data acquisition was performed using the Empower software(version 2.0).
A Dionex HPLC instrument was also used for intermediate precision measurements.The HPLC apparatus consisted of a Dionex P580 LPG pump,an ASI-100 T autosampler,an STH 585 column oven,and a PDA-100 UV detector;data were acquired and processed by a Chromeleon Data System(Dionex Corporation,Sunnyvale,California,US).
2.3.Preparation of system suitability and standard solutions
A system suitability solution was prepared by dissolving 2.0 mg of SRT-Peak-ID EP standard in a 20-mL volumetric flask with methanol.The solution was filtered through a 0.45-μm nylon membrane filter.In order to determine the SRT content,three standard solutions were prepared as follows:i)an accurately weighed amount of SRT,equivalent to about 50 mg of SRT free base,was transferred into a 100-mL volumetric flask;ii)after adding 75 mL of methanol,the resulting solution was maintained in an ultrasonic bath for 10 min and then diluted to volume with methanol;iii)the solution was diluted 2:20 and an aliquot was then filtered through a 0.45-μm nylon membrane filter(working concentration~50μg/mL).Suitable dilutions were carried out to obtain final concentrations of standard solutions from 50 to 0.5μg/mL.
2.4.Method validation
2.4.1.HPLC operating conditions
Analytical chromatographic separations were carried out on a Chiralpak IG-3 column(250 mm ×4.6 mm,3μm),with a mobile phase consisting of an acetonitrile-water-DEA 75:25:0.1(V/V/V)mixture at a flow rate of 1.0 mL/min;the column temperature was maintained at 30°C.The injection volume and sampler temperature were 10 μL and 5°C,respectively,and the detection wavelength was set at 215 or 246 nm for validation purposes.
2.4.2.Specificity
The selectivity of the analytical method was evaluated by analyzing the system suitability solution containing SRT and its related substances.Single standard solutions of each impurity were prepared by dissolving 1.0 mg of standard in 10 mL of methanol.
2.4.3.Linearity
The linearity was evaluated using standard solutions of SRT at concentrations ranging from 125 to 0.5μg/mL,with a working concentration of 50μg/mL.Three injections of each solution were made under the chromatographic conditions described above,using an injection volume of 10μL.The concentrations of the solutions were plotted against the corresponding peak area responses of SRT free base,and linear regression equations were then calculated.
Linearity tests for impurities were performed with a standard solution containing IMP-A,IMP-C,IMP-E,IMP-F,and IMP-G at concentrations ranging from 90-50 to 0.9-0.4μg/mL,with respect to an SRT working concentration of 1000μg/mL.
2.4.4.Limits of detection(LOD)and quantification(LOQ)
The LOD and LOQ parameters represent the concentrations of analyte that would yield S/N ratios of 3 and 10,respectively,according to the EP guidelines.The LOD and LOQ values of SRT and related impurities were determined by injecting a series of diluted solutions.
2.4.5.Precision
The precision of the method was determined by measuring the repeatability(intra-day)and intermediate precision(inter-day)of the retention times and peak areas measured for SRT and its impurities.The intra-day variability was measured by the same analyst over one day,while inter-day precision tests were carried out by another independent analyst,using a different HPLC apparatus over three days.The precision was determined by measuring the repeatability of the retention time and peak areas on replicate injections(n=6),and reported as percentage of relative standard deviation(RSD%).
2.5.Application of the proposed method
In order to determine the contents of SRT and its impurities in a commercial drug product,20 tablets containing 55.9 mg of SRT were accurately weighed and the average mass was calculated(155.91 mg).A portion of the ground tablet powder,corresponding to the average weight of two tablets,was transferred into a 100-mL volumetric flask,followed by the addition of 75 mL of methanol.After 10 min of sonication,the solution was diluted to volume with methanol and then stirred for 30 min.After centrifugation at 5000 rpm for 10 min,an aliquot was filtered through a 0.45-μm nylon membrane filter and analyzed by stereoselective HPLC.
3.Results and discussion
3.1.Enantio-,diastereo-,and chemo-selective reversed-phase HPLC analysis
In order to develop a suitable single-run HPLC method for the separation of SRT enantiomers in the presence of other impurities,we selected eluent mixtures composed of methanol-water-DEA and acetonitrile-water-DEA.DEA,as a basic additive,plays an important role in enhancing the symmetry of peaks and keeping the API and impurities as free bases.
Under such conditions,we explored the selectivity of a Chiralpak IG-3 column(250 mm×4.6 mm)with a methanol solution of SRT-Peak-ID,containing SRT spiked with the enantiomeric impurity IMP-G as well as with the IMP-A,IMP-B,IMP-C,and IMP-F impurities.The column temperature and flow rate were set at 25°C and 1 mL/min,respectively.The chromatograms obtained with the acetonitrile-water-DEA 90:10:0.1,80:20:0.1,and 75:25:0.1(V/V/V)mixtures are shown in Fig.2.As shown in the plots,a higher water content in the mobile phase resulted in a higher retention time of analyte(typical reversed-phase behavior)and a greater recognition ability ofthe Chiralpak IG-3 CSP.In particular,using the acetonitrile-water-DEA 75:25:0.1(V/V/V)mobile phase,a baseline separation of all chiral species was achieved within 18 min.Under these analytical conditions,the resolution between the SRT and IMP-G peaks was higher than 10,whereas that between the critical peak pairs SRT/IMP-B(i.e.,the second eluting enantiomer of IMP-B)and SRT/IMP-F(i.e.,the first eluting enantiomer of IMP-F)was higher than 2.
All species contained in the SRT-Peak-ID EP standard were identified by comparing the retention times(Fig.3)and UV absorption spectra with those of pure standards.The UV spectra recorded on-line during the chromatography run are shown in Fig.S1.As expected,the species having an enantiomeric relationship displayed the same UV profile.Nearly overlapping UV spectra were also observed for species having a diastereomeric relationship(SRT and IMP-A).
In an attempt to achieve a higher resolution,acetonitrile was replaced by methanol in the aqueous mobile phase used for the analysis of SRT in the presence of its related substances.The chromatographic profile displayed in Fig.3 indicates that the substitution of the organic modifier was unsuccessful,because a comparable overall resolution was not obtained.With a methanol content of 10%,the enantiomers of SRT were still well separated;however,the enantiomeric impurity(IMP-G)was closer to IMP-F compared to the chromatogram obtained with the optimized acetonitrile-based conditions(Rs=1.48 vs.2.02),and the second eluting enantiomer of IMP-B was co-eluted with SRT.Furthermore,the figure shows that the more retained enantiomer of the IMP-C was not separated from the other species.
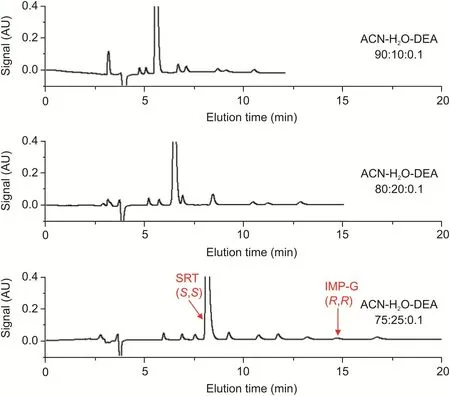
Fig.2.Typical HPLC chromatograms of SRT-Peak-ID EP standard using acetonitrile-water-DEA as mobile phase.Chromatographic conditions:column,Chiralpak IG-3(250 mm × 4.6 mm i.d.);mobile phase,as indicated in the figure;flow rate,1.0 mL/min;temperature,25°C;detection,UV at 215 nm.
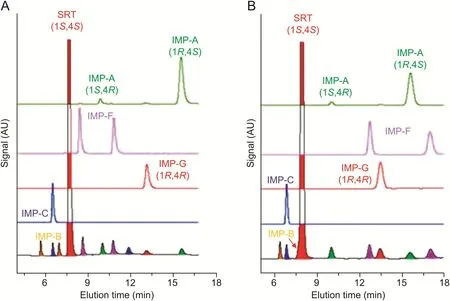
Fig.3.Comparison between HPLC chromatograms of SRT-Peak-ID EP standard and single impurities under reversed-phase mode.Chromatographic conditions:column,Chiralpak IG-3(250 mm × 4.6 mm i.d.);mobile phase,(A)acetonitrile-water-DEA 75:25:0.1(V/V/V)and(B)methanol-water-DEA 90:10:0.1(V/V/V);flow rate,1.0 mL/min;temperature,25°C;detection,UV at 215 nm.
3.2.Validation of the method
3.2.1.Specificity
As discussed above,the use of the acetonitrile-water-DEA 75:25:0.1(V/V/V)mixture as mobile phase enabled the baseline separation of SRT from its related substances.
To further improve the separation method,we investigated the influence of the column temperature on the performance of the Chiralpak IG-3 column.
By increasing the column temperature from 25 to 30°C,the analysis time reduced to about 14 min,whereas the resolution of the closely eluting peak pairs SRT/IMP-B and SRT/IMP-F was almost unchanged(Fig.4).Good peak efficiencies(60,000-80000 N/m)were recorded under these conditions.
Thus,the following experimental conditions were selected for the identification,characterization,and control of the SRT impurity levels:acetonitrile-water-DEA 75:25:0.1(V/V/V)as mobile phase,column temperature=30°C,and flow rate=1 mL/min.
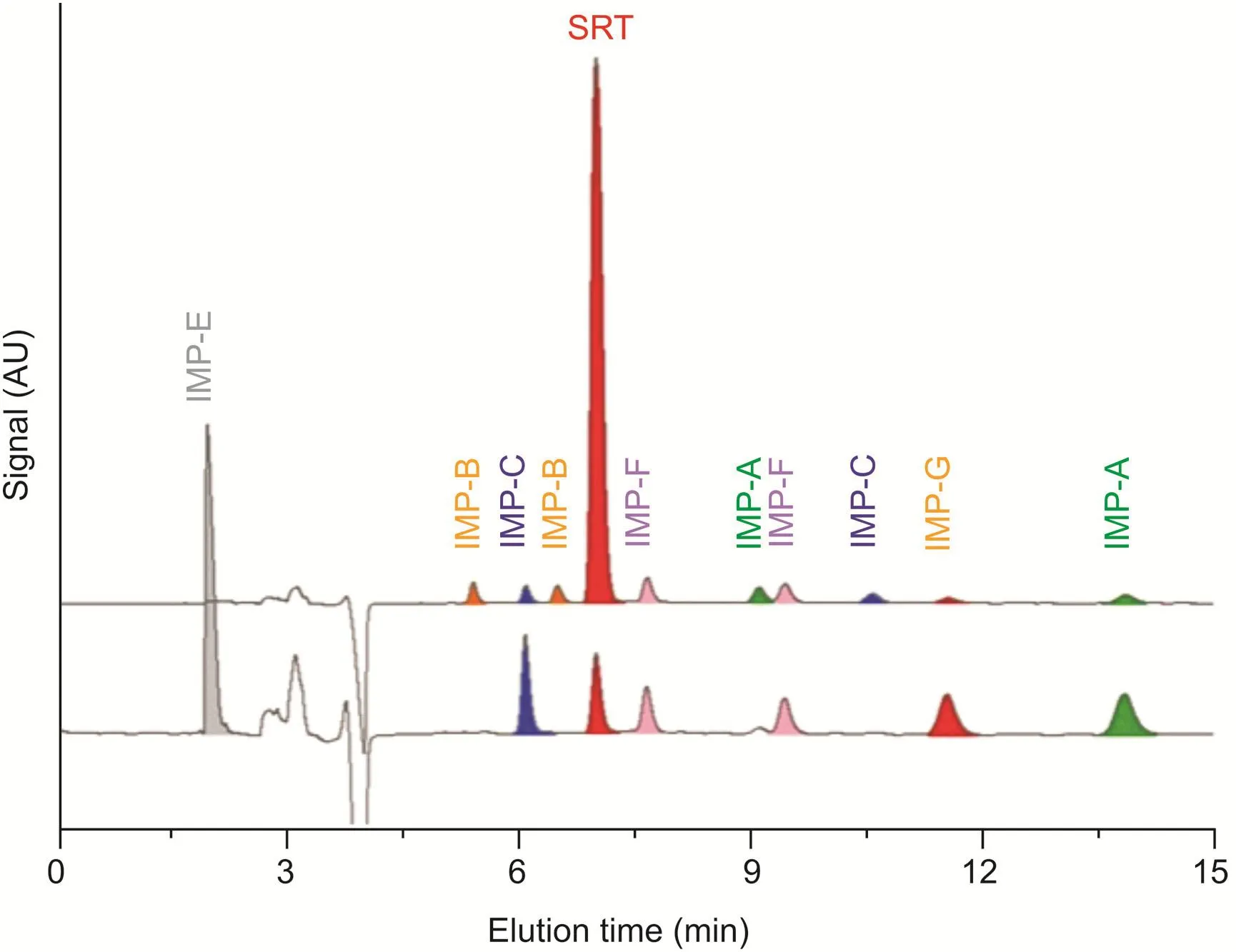
Fig.4.Comparison between HPLC chromatograms of SRT-Peak-ID EP standard(top)and reference sample containing SRT spiked with known impurities(bottom).Chromatographic conditions:column,Chiralpak IG-3(250 mm×4.6 mm i.d.);mobile phase,acetonitrile-water-DEA 75:25:0.1(V/V/V);flow rate,1.0 mL/min;temperature,30°C;detection,UV at 215 nm.
The next step was to evaluate the specificity of the optimized analytical method toward(R)-(-)-mandelic acid,which is used in the pharmaceutical synthetic industry as a chiral resolving agent for the isolation of SRT by preferential crystallization.This compound is considered as an additional impurity of SRT(named IMP-E in the EP monograph).As shown in Fig.4,IMP-E was unretained on the Chiralpak IG-3 column in the optimized aqueous conditions,and was eluted before column dead time.IMP-E is the only impurity with acidic properties;it is expected to be present as carboxylate anion in the presence of DEA in the mobile phase.Residual silanols are also expected to be partly deprotonated by DEA,resulting in ionic exclusion of IMP-E,thus partially excluding it from the column volume[18].Furthermore,the excipients used in the tablet manufacturing did not show any interference at the retention time of SRT and known impurities(chromatogram not shown).
It is worth noting that the peaks relative to the enantiomers of IMP-F in Fig.5 could be easily identified in the sample mixture by setting the wavelength at 246 nm,which corresponds to the selective wavelength of maximum absorbance.
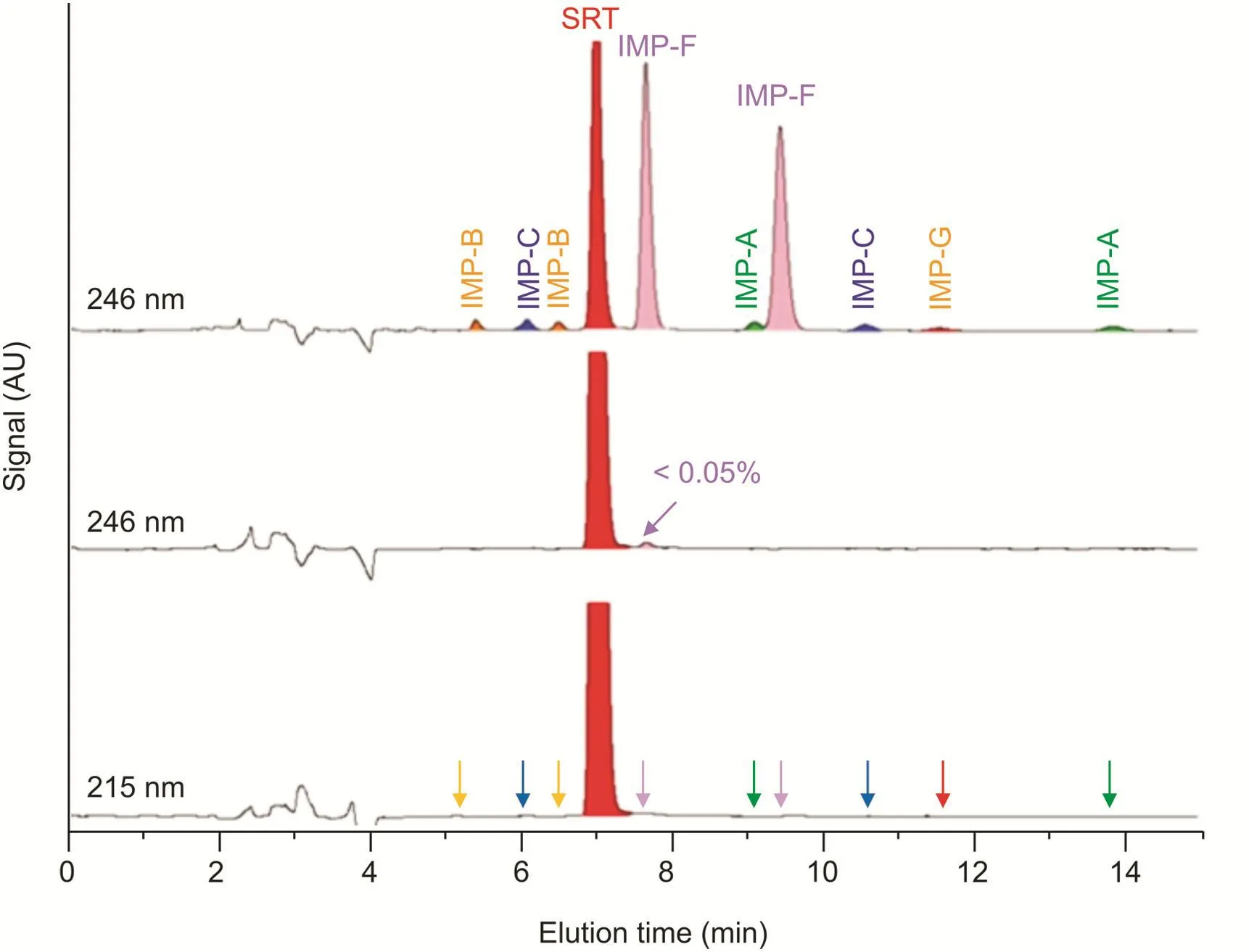
Fig.5.Comparison between HPLC chromatograms of SRT-Peak-ID sample with UV detection at 246 nm(top),solution of SRT free base tablet(50 mg)with UV detection at 246 nm(middle)and 215 nm(bottom).Chromatographic conditions:column,Chiralpak IG-3(250 mm×4.6 mm i.d.);mobile phase,acetonitrile-water-DEA 75:25:0.1(V/V/V);flow rate,1.0 mL/min;temperature,30°C.
Because the chlorophenyl Impurity D standard was not available,the specificity of the method toward this compound was not evaluated.
All the known impurities were found to be separated from each other and from the SRT peak,thus validating the HPLC method based on the immobilized Chiralpak IG-3 CSP;the results of further analyses are described as follows.
3.2.2.System suitability
The system suitability solution was used to determine the resolution between the critical peak pairs SRT/IMP-B and SRT/IMP-F.The resolution values were 2.11 and 2.67,respectively,which exceeded the limit parameter(Rs≥1.5).
The standard solutions for assay and related substances were also injected six times to evaluate the precision of the method;the results,reported in Table 1,are within the limit specifications.The obtained chromatograms are shown in Figs.S2 and S3.
3.2.3.Linearity
The linearity of the method was evaluated by injecting standard concentrations of SRT and impurity samples.The concentrations ranged from 0.5 to 124.6μg/mL(0.4%-150%)for SRT free base,0.8-7.9 μg/mL(0.08%-0.79%)for IMP-A free base,0.50-9.6 μg/mL(0.05%-0.96%)for IMP-C free base,0.35-70.0μg/mL(0.04%-7.0%)for IMP-E,0.4-7.8μg/mL(0.04%-0.78%)for IMP-F free base,and 0.8-8.2μg/mL(0.08%-0.82%)for IMP-G free base.
The peak area response was plotted against the nominal concentration(mg/mL)of the API or impurity.The linearity was evaluated by linear regression analysis,which was performed using the least squares regression method.The obtained calibration curve equations are shown in Table 1.
3.2.4.LOD and LOQ
The LOD and LOQ concentrations are shown in Table 1.S/N values of 3 and 10 were used as the criteria for the determination of the LOD and LOQ values,respectively.
3.2.5.Precision and repeatability
The precision of the method was determined by repeatability(intra-day)and intermediate precision(inter-day)tests.In the case of the SRT base,the method exhibited RSD% below 2.0% for both repeatability(0.02% for retention times and 0.54% for peak areas)and intermediate precision(0.03% for retention times and 0.61% for peak areas),which met the proposed acceptance criteria(RSD% not higher than 2.0%).
In the case of impurities,the method gave the following RSD% for retention times and peak areas:IMP-A free base,0.08%-2.8%(repeatability)and 0.09%-3.5%(intermediate precision);IMP-C free base,0.08%-2.2%(repeatability)and 0.09%-2.5%(intermediate precision);IMP-E,0.08%-2.1%(repeatability)and 0.08%-2.2%(intermediate precision);IMP-F free base,0.09%-1.9%(repeatability)and 0.08%-2.1%(intermediate precision);IMP-G free base,0.09%-1.4%(repeatability)and 0.08%-1.6%(intermediate precision).The repeatability and intermediate precision data met the proposed acceptance criteria(RSD% not higher than 5.0%).
3.2.6.Accuracy
Further analyses were carried out to evaluate the recovery of SRT and known impurities.The accuracy of the determination of enantiopure SRT was determined by preparing three drug samples at 50%,75%,and 150% of the target(50μg/mL).The apparent recovery ranged from 99.1% to 99.9%.
The accuracy of the determination of each impurity was determined by preparing two samples at 0.3% and 0.5% of the target(1000μg/mL).The apparent recovery ranged from 95.1% to 97.2%.
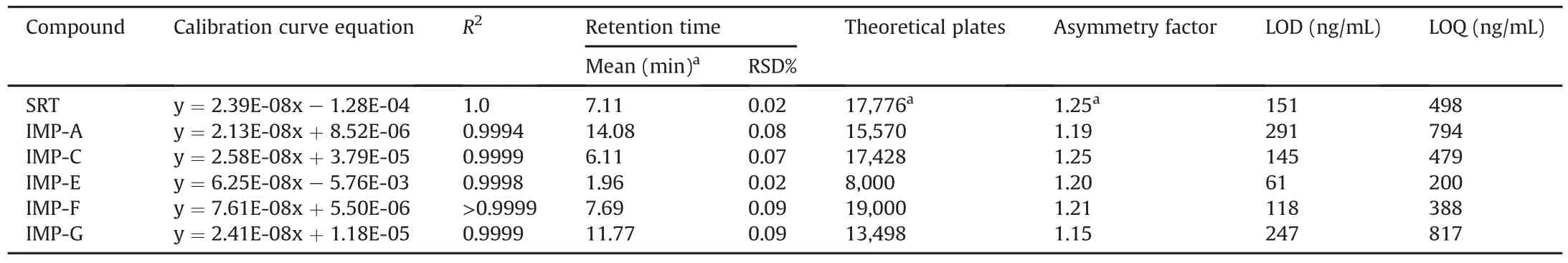
Table 1Results of system suitability and linearity tests(calibration curve equation,detection limit and quantification limit).
3.2.7.Robustness
Another set of experiments was carried out to evaluate the effects of variations in flow rate,column temperature,and mobile phase composition.For this purpose,the system suitability solution was analyzed at flow rates of 0.9 and 1.1 mL/min and temperatures of 28 and 32°C,using acetonitrile-water-DEA 75:25:0.1(V/V/V)±1% mixtures of each component as mobile phases.The results were found to lie within acceptable limits,and the relative retention times of known impurities were not significantly different from those of the reference conditions.
3.3.Determination of SRT and impurity contents in commercial drug product
The final step of our study was to analyze a marketed formulation(tablets)containing 55.9mg of SRT,corresponding to 50.0mg of SRT free base,using the optimized method based on the Chiralpak IG-3 CSP.Typical chromatograms obtained from the analysis of test solutions are shown in Fig.5.The comparison of the HPLC profiles obtained by setting the UV detector wavelength at 246 and 215 nm shows that only the peak corresponding to IMP-F could be detected and quantified(i.e.,<0.05%).In order to determine the content of SRT base in the tablets,the three test solutions were diluted 1:20 and analyzed in the same conditions used for the determination of related substances.The average measured content was 100.9%,corresponding to 50.5 mg of SRT free base(RSD%=0.9).
4.Conclusions
In summary,a simple and rapid HPLC method using the Chiralpak IG-3 column in reversed-phase mode was developed for the simultaneous assay and related substance determination of SRT samples.The proposed method,which does not use buffers or tandem-coupled columns,is selective,precise,accurate,and meets the requirements set in the EP guidelines.
Furthermore,we demonstrated the applicability of the singlerun chiral approach for the routine control of the content of SRT and its related substances in commercial tablets.
Therefore,the described procedure could represent a useful and valuable tool for reducing economic and environmental costs in the analysis of SRT-based raw materials,working standards,and finished product on an industrial scale.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
On the occasion of her retirement,the authors wish to dedicate this work to Dr.Luisa Valvo.
The authors are grateful to Ms.Antonina Mosca(Istituto Superiore di Sanità)for her technical assistance and Dr.Paolo Vatta(Istituto Superiore di Sanità)for improving the English language of the manuscript.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.11.002.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Challenges for cysteamine stabilization,quantification,and biological effects improvement
- Offline two-dimensional liquid chromatography coupled with ion mobility-quadrupole time-of-flight mass spectrometry enabling fourdimensional separation and characterization of the multicomponents from white ginseng and red ginseng
- Development of a UHPLC-MS/MS method for the quantification of ilaprazole enantiomers in rat plasma and its pharmacokinetic application
- Use of subcutaneous tocilizumab to prepare intravenous solutions for COVID-19 emergency shortage:Comparative analytical study of physicochemical quality attributes
- Discovery of human coronaviruses pan-papain-like protease inhibitors using computational approaches
- Vancomycin pretreatment attenuates acetaminophen-induced liver injury through 2-hydroxybutyric acid