基于非天然结构组件的人工酶设计与应用
2021-01-21袁飞燕于洋李春
袁飞燕,于洋,李春
(北京理工大学化学与化工学院生化工程系,合成生物系统研究所,北京 100081)
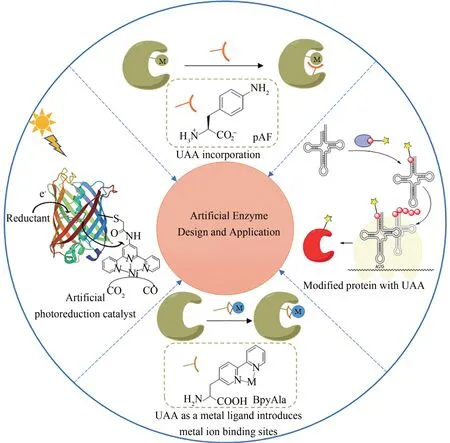
酶的高催化性能促进其在化学品合成和材料催化剂中的应用。部分天然酶存在成本昂贵、产率低、稳定性差等不足。在参与氧化还原反应的酶中,多数酶在反应中需要辅酶或辅基[1],这更加大了天然酶的使用成本。人工酶是以酶的催化原理为基础,模拟酶的生物催化功能,用化学法和生物法合成的具有特定催化功能的生物大分子。人工酶可以弥补天然酶的不足。人工酶已被用作控制化学反应的高效催化剂[2],同时有助于天然酶机理的深入研究[3]。此外,天然酶对底物具有高度专一性而使底物种类受限,影响在工业上的应用,而人工酶可扩大反应底物范围[4]。
在对天然酶的研究和改造中,研究者发展出了定向进化、理性设计等方法。例如在定向进化中常用到的易错聚合酶链反应(epPCR)和DNA-shuffling。通过在酶的结合口袋内分区饱和诱变也成为一种有效的方法,称为组合活性中心饱和测试(CAST)。而迭代饱和突变(ⅠSM)更适合进一步的遗传优化[5]。通过改变氨基酸序列对酶进行改造,得到具有特定功能的酶,成为众多研究者青睐的方法。研究者为了创造新的、具有与天然酶催化能力相当,甚至优于天然酶催化能力的人工催化剂,常常对酶的底物识别位点、活性位点催化基团以及疏水微环境进行设计[6-7]。Arnold等[8-19]从具有单加氧活性的P450 酶出发,通过定向进化改变氨基酸序列,使P450酶可以催化C—H活化和C—C、C—N、C—S、C—Si 键形成,提高其催化效率或产生新的底物特异性,实现了天然酶不能催化,甚至化学方法难以进行的反应。但是蛋白质是由20 种天然氨基酸残基(此外还有硒代半胱氨酸和吡咯赖氨酸)以及有限种类的天然辅因子组成,这大大限制了蛋白质可实现的结构、反应性、功能空间。而非天然结构组件(包括非天然氨基酸及非天然辅因子)的引入,可以将一些特殊化学基团(如叠氮基、炔基、联吡啶等)掺入蛋白质特定位点,或引入具有荧光等特殊光谱学性质、模拟翻译后修饰等的氨基酸,这都可以为拓展人工酶的性质提供新的科学视角。因此,引入非天然结构组件构建人工酶,已成为新的人工酶的设计策略。本文作者将以参与氧化还原反应的人工金属酶为例,探讨基于非天然结构组件的人工酶的设计方法。展望引入非天然结构组件结合计算设计或代谢工程构建人工酶的新方法,这有助于实现媲美天然酶效率的人工酶的设计与应用。
1 非天然氨基酸的引入策略
利用非天然氨基酸(UAAs)扩展遗传密码大大增加了蛋白质可供改造的化学空间。基因密码子扩展技术[20]是利用无义密码子实现蛋白质中非天然氨基酸的定点引入的方法。这种方法的关键是保持正交性。具体而言,UAA 的密码子不应编码共同的氨基酸;新的tRNA/氨酰tRNA 合成酶(aaRS)都不与任何内源性tRNA 合成酶对发生反应;新的合成酶只应识别UAA,保证不识别20 种天然氨基酸[21]。正交 aaRS 将 UAA 装载到 tRNA上,该tRNA 在核糖体中识别mRNA 上相应的无义密码子,从而导致UAA 掺入目标蛋白中。将具有叠氮、炔基等活性基团的UAA 掺入蛋白质中,可以利用Cu(Ⅰ)催化的叠氮化物-炔烃环加成反应偶联小分子催化剂[图1(a)][22-23]。这一技术可以在蛋白质特定位点引入具有特殊功能或化学性质的非天然氨基酸,为拓展蛋白质的性质或者创造全新的蛋白质提供了强有力的工具。
金属是酶催化中的重要辅因子,在蛋白质结构中起着关键的调节作用[24]。金属酶可以催化氧化还原、水解等多种反应,参与了生物圈重要的物质转化过程。但是,在天然氨基酸中,仅有7种的侧链可以与金属离子配位,而金属离子结合位点需要精确定位多个氨基酸配体,这使得设计全新的金属结合位点,构建人工酶成为巨大的挑战。小分子配位化学研究中有很多常用的多齿配体,在一个分子内通过不同的官能团与金属离子形成配位作用,对金属离子有很高的亲和力。我们可以在蛋白质中方便地通过引入含多齿配体侧链的非天然氨基酸构建高亲和力的金属位点,实现人工酶的设计[图 1(b)][25]。例如 Liu 等[26]在绿色荧光蛋白中引入8-羟基喹啉丙氨酸(HqAla),可以得到对铜的亲和力为fmol/L 级别的金属结合位点,这一亲和力已经高于很多天然的金属酶。
非天然氨基酸可以用于人工金属酶的构建。大多数的过渡金属催化的反应不能由天然金属酶介导,而引入非天然氨基酸则允许过渡金属的位点特异性配位,从而形成新的人工金属酶。LmrR是一个细菌的转录抑制蛋白,其二聚体间含有一个大的疏水孔,它杂泛的结合口袋为平面分子结合提供了一个合适的环境。Drienovská 等将LmrR的基因中M89 位突变为对应琥珀终止密码子的TAG,用于非天然氨基酸引入。M89 位于疏水口袋的远端,从而避免了金属配合物形成活性差的2:1 配体的风险。通过将非天然氨基酸(2,2'-联吡啶-5基)丙氨酸(BpyAla)引入LmrR,提供一个稳定的金属结合位点,使得Cu(Ⅱ)能被结合到BpyAla 上。他们通过遗传方法快速优化构建人工金属酶LmrR_LM_M89X_F93W(其中X 为非天然氨基酸BpyAla),可催化不对称Friedel-Crafts 烷基化反应。在该人工酶催化的不对称乙烯基Friedel-Crafts烷基化反应中,产物的ee值高达83%[27-28]。
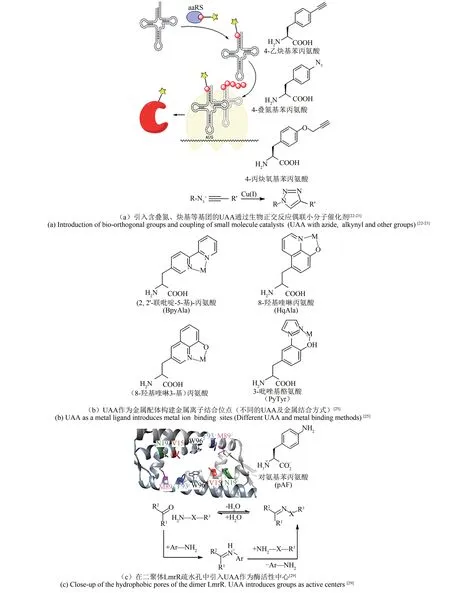
图 1 利用 UAAs构建人工酶的设计策略[22-23,25,29]Fig.1 Design strategies for constructing artificial enzymes using UAAs[22-23,25,29]
除了通过引入金属离子外,非天然氨基酸中的独特官能团还可以在活性中心直接参与反应。Drienovská等[29]通过引入对叠氮苯丙氨酸(pAzF)还原的策略将非天然氨基酸引入至LmrR 结合口袋附近的四个不同的位置(Ⅴ15、N19、M89 和F93)作为酶活性中心,构建了新的人工酶[图1(c)][29]。他们首先在LmrR 中引入 pAzF,再用三羧基乙基膦(TCEP)将其还原成对氨基苯丙氨酸(pAF)。这解决了直接引入pAF导致的低表达率及酪氨酸和苯丙氨酸的错误掺入的问题。构建的人工酶中LmrR_Ⅴ15pAF 具有独特的苯胺侧链,增加了酶的反应活性,可以作为形成腙的良好催化剂(产率为 72% ± 3%)。UAA 引入的 pAF 作为活性反应中心,在苯胺的存在下形成亚胺离子中间体,加速了腙(X=NH)和肟(X=O)的形成[图1(c)][29]。在此基础上,最近Mayer等[30]将以非天然氨基酸为催化残基的人工酶LmrR_Ⅴ15pAF 作为定向进化的起点,通过识别蛋白质支架中的有益突变,增强非天然侧链的固有催化活性,使酶的kcat增 加 至 452 s-1, 相 比 LmrR_Ⅴ15pAF 提 高 了 近100倍,促进了腙的高效合成。
2 非天然辅因子的引入策略
天然酶常使用卟啉、钴胺素等金属配合物作为辅因子参与催化反应。天然酶使用的金属局限在铁、铜、锌、钴等少数种类,配体的类型也很单一。随着金属有机催化的发展,很多过渡金属,尤其是贵金属被认为具有很好的催化活性。将这类金属配合物作为辅因子与蛋白质结合,可以构建人工酶,催化新的反应。
链霉亲和素(SAⅤ)已被广泛用作制造人工金属酶的支架。Whitesides 等[31]在亲和素中引入了一种生物素化的有机金属催化剂,以提供一种混合催化剂,该催化剂结合了酶和有机金属催化剂的特征。生物素化的有机金属辅因子对链霉亲和素的非共价亲和力提供了良好的稳定性,将少量的生物素化有机金属催化剂与SAⅤ突变体结合起来,可以产生显著的多样性,从而优化人工金属酶的催化性能[图 2(a)][32-33]。Jeschek 等[34]证明了链霉亲和素生物素技术在体内的适用性的同时,用此策略创造人工金属酶催化天然酶中不存在的一种反应机制——烯烃复分解反应。此方法得到的人工金属酶还可以通过针对不同底物的定向进化继续改进人工酶的活性。
以血红素作为辅因子的蛋白参与氧气运输或氧激活反应,是金属酶研究的焦点之一[35-37]。其中,肌红蛋白和血红蛋白结构简单,易于获取,被公认为分子工程研究的优良起点[38]。在20 世纪上半叶就有研究者通过将蛋白脱辅基、重组在血红蛋白中引入非天然卟啉[39]。研究人员分别在肌红蛋白中引入非天然卟啉,构建了不同的人工酶[40-43]。其中,Lu课题组用非天然血红素和非天然金属离子替代天然的血红素及金属离子,以调节底物亲和力、电子转移速率和酶的活性。例如将非生物辅因子锰-双席夫碱复合物掺入肌红蛋白中,赋予其生物学功能,并证明了金属因子的锚定可以调节酶的对映选择性。此外,他们还探讨了蛋白质主链的相互作用;证明了疏水性和H键网络的重要性;以及还原电势是轴向配体疏水性主要决定因素[43-45]。
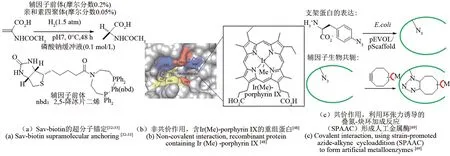
图2 非天然辅因子不同的引入策略[32-33,48,69]Fig.2 Different introduction strategies for non-natural cofactors[32-33,48,69]
Hartwig 课题组将金属有机催化的研究经验结合到人工金属酶的构建,在蛋白质中加入了完整的贵金属复合物,从而产生了人造金属蛋白。他们将有机金属配合物的反应多样性与酶的选择性相结合,构建出具有类似天然酶活性的人工酶[46]。Key 等[47]将肌红蛋白中铁卟啉(Fe-porphyrin ⅠX)替换为含有 Fe(Cl),Co(Cl),Cu,Mn(Cl),Rh,Ⅰr(Cl),Ⅰr(Me),Ru(CO)和Ag的卟啉结构。其中含有 Ⅰr(Me)-porphyrin ⅠX 的重组蛋白[图2(b)][48]显示出对卡宾转移反应具有一定的催化能力。
含 Ⅰr(Me)-porphyrin ⅠX 的重组肌红蛋白较天然酶的催化速率还相差甚远,所以Dydio 等[46]选择了热稳定性更好的P450酶CYP119,将铁卟啉替换 为 Ⅰr(Me) -PⅠX。 所 得 到 的 人 工 金 属 酶 Ⅰr(Me)-porphyrin ⅠX CYP119 经过定向进化,反应速率和选择性大大提高,Ⅰr(Me)-porphyrin ⅠX CYP119 的突变体催化卡宾插入C—H 键,其ee 值高达98%。最终得到的人工酶催化卡宾插入C—H键的TOF 达到2550 h-1,总转化数35 000,达到了与天然P450酶催化单加氧反应相似的水平。Dydio等[49]证明了 Ⅰr(Me)-CYP119 酶催化 C—H 胺化反应具有较高的化学选择性,可用于插入硝基苯还原为磺胺。Ⅰr(Me)P450 催化的插入反应容纳了小分子催化剂不能容纳的一系列官能团,其底物范围比含铁卟啉的酶更广[50]。以上Hartwig课题组提出的人工金属蛋白的制备方法,为多种卟啉-蛋白质支架和非天然金属辅因子合成人工酶催化的广泛的非生物转化奠定了基础。
改造天然辅因子得到的新的辅因子也可以为新的酶化学提供助力。Ji 等[51]合成了三种新的烟酰胺腺嘌呤二核苷酸(NAD)类似物。F作为一个强吸电子基团在C5位置损害辅因子结合特性,因此用Cl、Br 或甲基取代F 合成三种新的NAD 类似物。就它们催化L-苹果酸的氧化脱羧而言,每对NAD 类似物和双突变体(ME L310R/Q401C)均与NAD 和天然大肠杆菌苹果酸酶ME 具有良好的正交性,可以进一步探索氧化还原酶与辅因子之间的分子相互作用,从而产生新的生物正交氧化还原体系。
除了简单的金属离子、金属配位化合物外,一些酶含有金属团簇,如固氮酶中的铁钼辅因子、光系统中的光合放氧中心等,催化了许多重要的反应[52-57]。在蛋白质骨架中引入这些团簇是对蛋白质设计的挑战。Mirts 等[58-62]选择分子量小且稳定的细胞色素c 过氧化物酶(CcP)作为模型,其活性位点有一个足以容纳[4Fe-4S]簇的空腔。通过体外重组在Apo-SiRCcP 中加入了一个铁硫簇,设计了多核人工金属酶用作催化剂。理性设计金属和底物结合位点周围的第二配位层,三个Cys突变(T180C,W191C,L232C)以协调[4Fe-4S],稳定性突变M230A 用于缓解空间碰撞,D235Ⅴ用以移除干扰CcP 中的Cys-Heme 协调附近的负电荷,H175C突变作为血红素与[4Fe-4S]辅因子之间的桥联Cys配体,使其形成了紧密的结构,这一系列理性设计创造了接近天然酶SiR 活性的人工金属酶,这些第二配位相互作用对于优化设计策略以实现人工酶的多电子氧化还原的高活性具有重要意义。此种策略为设计多种人工金属酶提供了机会,甚至超过小分子催化剂的化学反应活性。
除了直接改造辅因子,对底物结合口袋的改造也可以实现人工酶的构建,比如Watanabe 小组发展的诱饵分子(decoy molecule)策略。一种模式P450 酶,P450 BM3 通常以长链脂肪酸为底物。Watanabe 等通过引入诱饵分子结合在活性口袋中构建特定的底物结合位点,极大地增强了P450 酶对小分子非天然底物的催化杂泛性[63-64],实现了丙烷、苯等非天然底物的羟化。研究人员将该策略用于小分子底物光脱羧酶[65]和烯烃水合酶[66]的构建,显示了这一策略在人工酶设计方面的潜力。Cong 等[67-68]报道了双功能小分子(dual-functional small molecule,DFSM)策略,令诱饵分子的酰基氨基酸基团与酶的活性口袋结合,而咪唑基在H2O2的活化中发挥酸碱催化剂的作用,成功将P450 单加氧酶改造为过加氧酶(peroxygenase),摆脱了对NADPH辅酶的依赖。该研究拓展了P450酶在有机合成中的潜在用途,为人工P450 酶的设计提供了新的途径。
3 非天然氨基酸与非天然辅因子的协同引入策略
非天然氨基酸和非天然辅因子的引入可以大大扩展蛋白质结构中可及的官能团,拓展现有酶的功能。而同时引入非天然氨基酸和非天然辅因子,可以进一步增加人们对蛋白质的改造能力,得到定制化的人工酶。
相比通过非共价相互作用引入非天然辅因子,将非天然辅因子与骨架蛋白共价偶联可以提高人工酶的鲁棒性。通过基因密码子扩展引入含生物正交基团的非天然氨基酸,可以实现非天然辅因子的定点特异共价锚定。利用环张力诱导的叠氮-炔环加成反应(SPAAC)将金属催化剂引入蛋白质,形成人工金属酶,同时消除自然发生的锚定残基带来的选择性、反应性差等限制[图2(c)][69]。Yang等[69]通过pAzF将含铑配体引入咪唑甘油磷酸合酶的合成酶亚基(tHisF)中,实现了卡宾转移和C—C、C—Si键成键。
化学家一直致力于设计催化剂,以模仿自然光合系统获取可见光光子的能力,并利用光子吸收的能量来驱动化学反应。Liu等[70]设计了一种光催化CO2还原酶。他们在黄色荧光蛋白(sfYFP)中,使用遗传密码子扩展技术用二苯甲酮-丙氨酸(BpA)替换发色团残基Tyr66,而Gly65-BpA66-Gly67可自催化转化为高荧光的对羟基苄基-5-咪唑啉酮的发色团,从而设计得到一种新的光敏蛋白(PSP),激发后转换为能够用于较远距离电子转移的三重态,氧化弱牺牲还原剂产生一个强还原自由基,用于驱动CO2还原催化剂的还原。为了将PSP转化为光催化CO2还原酶,他们使用了三联吡啶镍配合物,成功进行了有效的光化学还原反应,催化CO2还原为CO(图3)[70]。该策略代表了一种很有前景的光氧化还原酶设计的新方法,同时也为具有非天然光催化活性的人工酶提供了新的方向。
4 人工酶的高效制备策略
由于翻译效率的降低,含UAA 的蛋白质的产率往往比野生型蛋白质的产率低,但某些非天然氨基酸可以有较好的产率。Schultz 等[71]使用tRNACUA
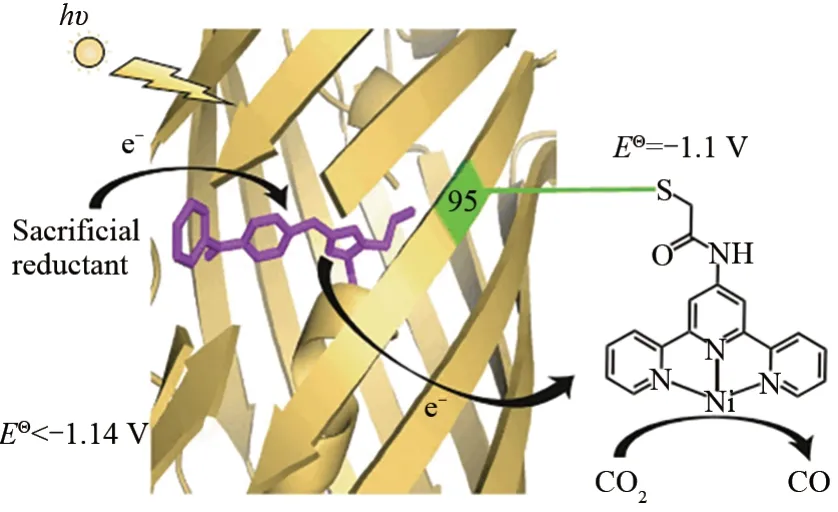
图3 PSP2T催化机理示意图[70]Fig.3 Schematic diagram of the proposed catalytic mechanism of PSP2T[70]
Tyr/MjTyrRS 正交对将非天然氨基酸pAcF 分别引入T4溶菌酶的第68位和酮类固醇异构酶的78位,在这两种情况下都观察到有效的蛋白质表达,其产率相应的约为野生型蛋白的50%。而对碘苯丙氨酸(pⅠPhe)、二氟代酪氨酸(F2Tyr)的掺入已经在一系列蛋白质展现出接近野生型的产量[72-74]。
由于非天然氨基酸和非天然辅因子大部分时候需要通过化学合成,导致人工酶成本较高、难以直接在细胞中直接制备。通过酶工程与代谢工程相结合,可以在细胞内构建非天然氨基酸和非天然辅因子的合成通路,实现人工酶的体内组装并直接参与细胞中的代谢反应。在大肠杆菌中加入生物合成途径以及正交的tRNA 合成酶-tRNA对,产生新的大肠杆菌表达系统,可在体内掺入非天然氨基酸,如 pAF[75]和吡咯氨基酸(Pyl)[76]。pAF 是氯霉素等天然产物合成的中间体。Mehl等[75]在大肠杆菌中引入Streptomyces venezuelae强化分支酸的合成。过量积累的分支酸被大肠杆菌内源的酪氨酸转氨酶转化生成pAF。经过代谢强化,内源生产的pAF 可以被相应的tRNA 合成酶识别,引入到蛋白质中。
Marchand 等[77]确定并表征了一种生物合成途径,用于生产含有卤素、末端烯烃和末端炔烃的氨基酸,并采用了一种氨酰-tRNA合成酶(PraRS)取代Met残基的方法,证明这些内源产生的氨基酸可以翻译成蛋白质。这些非天然氨基酸的官能团被引入肽和蛋白质的特定位点为多种下游生物正交化学提供了机会。特别是末端炔基通过Cu(Ⅰ)催化叠氮-炔环加成“点击”反应(CuAAC)得以被广泛应用。类似地,非天然氨基酸氟硫酸盐-酪氨酸(FSY)被成功引入蛋白质中,利用基因编码的化学转化策略(GECCO),通过硫氟化交换(SUFEX)选择性与近端氨基酸反应,在活细胞蛋白质中引入新的化学反应赋予新的共价键能力[78-79]。
5 总结与展望
在人工酶设计的过程中,无论采取哪种策略都不可避免地存在耗时费力的壁垒,特别是想将蛋白质催化剂的效率提高几个数量级。而理性设计人工酶,引入UAAs等非天然结构组件选择正确的特定位点也是一个挑战。很多通过数据库检索或者组学分析得到的蛋白质往往没有三维结构,同源建模的方法可以基于氨基酸或者核酸序列预测蛋白质的结构。通过计算机辅助或者人工智能可以大大降低人工酶设计的工作量。量子力学/分子力学(QM/MM)模拟计算方法已被用来优化人工酶设计及阐明其机制[80]。Rosseta 等软件基于大数据中得到的能量函数对蛋白质结构进行预测,寻找低能量构像,这种设计方法也被应用到含有非天然氨基酸的酶催化剂的设计中。同时,Mills等[81]将Rosetta设计方法用于设计金属蛋白,利用Bpy-Ala 作为结合金属离子的主要配体。他们设计了一个金属结合位点,由Bpy-Ala、两个蛋白质金属配体以及两种金属结合水分子组成的八面体配位几何结构。Bpy-Ala介导的金属蛋白具有与Co2+、Zn2+、Fe2+和Ni2+等二价阳离子结合的能力。同时,Mills 团队对设计的蛋白质进行X 射线晶体分析,发现晶体结构与计算设计的模型非常接近,展示了Rosetta对于金属蛋白设计的能力。Yang等[82]最近对18 个蛋白的从头设计,更进一步证明了Rosetta 可以更精确地设计蛋白模型。计算方法的改进,为人工酶的计算设计和实验室进化提供了良好的起点[83-84]。
综上所述,通过引入非天然氨基酸及非天然辅辅因子等多种策略能有效合成具有新功能和新结构性质的人工酶。有机金属的合理引入也为人工酶的设计开发了更多的催化性能。非天然氨基酸代谢合成系统的创造,也可用于体内直接合成非天然氨基酸,引入多肽药物中具有新药效的结构,进一步促进生物医药、绿色化学等行业的发展。正交氨酰tRNA 合成酶可以在表达宿主体内将非天然氨基酸特异性地引入蛋白的特定位点,生产修饰蛋白可以避免人工添加非天然氨基酸的烦琐,也能降低生产成本。非天然结构组件与蛋白质的结合为新反应的拓展提供了基础。
目前为止,大部分人工酶只能在纯化后进行反应。随着合成生物学的发展,将具有非天然组件的人工酶与细胞代谢过程进行整合,合成自然界不存在的分子也成为一种前沿课题。Reynolds等改造细胞色素P450,利用空间位阻使其识别小于血红素的卟啉分子,又引入血红素[85]转运通道ChuA,向细胞内导入非天然卟啉。他们构建了正交酶/辅因子对,可能为增加辅因子多样性和扩大细胞蛋白质化学功能提供一种通用策略,使辅因子选择性地结合到正交蛋白支架,而不与内源性元件发生干扰反应。在一篇ChemRxiv 预印本论文中,Huang 等[86]通过与 ChuA 类似的 hug 作为卟啉转运体系,将铱卟啉转运进入大肠杆菌细胞中。在大肠杆菌细胞中表达的CYP119 突变体可以结合铱卟啉,催化了以香芹酮为底物的卡宾转移反应,合成了一种非天然的萜类化合物。将人工酶与代谢工程结合,可能合成大量的“非天然”的天然产物,拓展现有的分子结构空间。
非天然结构组件的合理利用为快速进化人工酶、建立具有全新功能的平台奠定了基础。纵然应用非天然结构组件的引入策略得到了一些新的人工酶,但实际工作中,高效的人工酶设计往往需要定向进化、计算模拟甚至代谢工程的结合,动力学因素可能是催化活性的关键。这种多元化方法的结合有望推动更多与天然酶催化效率相媲美的人工酶的发现。
