Ce掺杂的NiCo2O4催化氧化甲苯性能研究
2021-01-14王洪华刘浩彬牛建瑞
王洪华,刘浩彬,牛建瑞*
(1.河北省环境科学研究院,石家庄 050000;2.河北科技大学环境科学与工程学院,石家庄 050018)
工业快速发展过程中排放大量的甲苯容易让人产生恶心和头痛等不适症状,且长期接触可引起慢性中毒及神经系统和肝脏损伤,甚至有致癌的风险,严重影响了人类的生活[1]。因此寻求绿色、安全、无二次污染技术是解决甲苯问题的关键。与此同时,催化氧化技术可彻底矿化甲苯而被广泛应用于甲苯处理中[2]。
催化氧化技术的核心是催化剂[3]。目前,催化材料有贵金属、非贵金属氧化物、复合型非贵金属氧化物(包括钙钛矿型和尖晶石型等[4])。其中,Co3O4具有较好的催化氧化活性,是很有前途的VOCs催化剂。基于此研究开发了多种类型的 Co3O4催化剂,包括纳米线和纳米棒催化剂[5]、有序介孔Co3O4催化剂、三维有序纳米Co3O4催化剂[6-8]和负载型催化剂(如以CNTs、Beta、ZSM-5、SBA-15、介孔硅为载体的催化剂[9-10])。以上CO3O4材料在催化降解VOCs上都表现出了非常优异的活性。众所周知,较大比表面积、丰富的表面吸附氧、催化剂表面较高浓度的高价态金属离子及较多的表面缺陷可提升催化材料活性[11],然而此类单金属氧化物催化剂只能通过改变其物理结构来增强催化性能,且表面性质调控难以实现。
通过金属掺杂形成双金属氧化物被认为是一种可调控材料表面性质的方法之一。其中通过金属掺杂后形成尖晶石催化剂,此催化剂具有起燃温度低、廉价、无二次污染等优势,逐渐成为催化降解甲苯的主流催化剂。Huang等[12]研发了一种新型、低成本、高性能的3D-NiCo2O4纳米片状催化剂,用于室温下氧化甲醛。此催化剂比表面积大,吸附有机物能力强,显示出很高的催化氧化活性,室温下甲醛的转化效率可达95.3%,且连续使用200 h后催化性能基本不变。Tao等[13]采用共沉淀和热分解法合成了NiCo2O4、Ni1.25Co1.75O4、Ni0.75Co2.25O4纳米粒子,其中NiCo2O4显示出最高的活性,在350~550 ℃温度范围内可完全降解甲烷。通过以上研究可以看出,NiCo2O4尖晶石在催化氧化等方面性能优异。然而,目前催化氧化性能优越的NiCo2O4[14]尖晶石多应用在电化学上,在甲苯氧化上的应用较少。强化催化剂表面活性氧生成速率有利于进一步提升其催化性能。CeO2具有较好的储-放氧能力,能与过渡金属之间存在特定的电子交换,且掺杂铈可提升催化剂高价态金属含量,改变催化材料的氧化还原性质,有利于加快催化剂表面活性氧的产生速率[15]。Wang等[16]采用Al/Ce和Al/Zr共掺杂Fe2O3催化剂,用于煤焦油的蒸汽催化裂化,掺杂后提高了轻质煤油的收率,光电子能谱(X-ray photoelectron,XPS)分析,掺杂可以增加O-的浓度,结果表明O-是决定蒸汽催化裂解的主要因素之一。
现采用水热法制备Ce掺杂的NiCo2O4尖晶石催化剂,以甲苯为目标污染物,考察掺杂方式和掺杂量对降解甲苯效果的影响,并深入探讨催化剂的稳定性及其催化甲苯机理,以期为催化氧化技术的工业化应用提供技术支持。
1 实验部分
1.1 催化剂制备方法
1.1.1 一次水热法制备NiCo2O4催化剂
把1 mol/L Na2CO3溶液5 mL、无水乙醇12.5 mL、1 mol/L Co(NO3)2·6H2O溶液3.4 mL、氨水11.5 mL、1 mol/L Ni(NO3)2·6H2O溶液1.7 mL,在集热式恒温加热磁力搅拌器剧烈搅拌下以2 min的间隔分别加入,将前体溶液再搅拌20 min,将酒红色均匀溶液转移至高压反应釜内,高压反应釜在鼓风干燥箱中170 ℃水热17 h;洗涤抽滤,在干燥箱中80 ℃下烘干200 min,烘干后在马弗炉空气氛围中,以3 ℃/min升温至300 ℃焙烧200 min得到目标催化剂。
1.1.2 一次水热法制备Ce掺杂NiCo2O4催化剂
将1 mol/L Na2CO3溶液5 mL、无水乙醇12.5 mL、1 mol/L Co(NO3)2·6H2O溶液3.4 mL、氨水11.5 mL、1 mol/L Ni(NO3)2·6H2O溶液1.7 mL,Ce(NO3)3·6H2O[物质的量比为Ce(NO3)3·6H2O∶Co(NO3)2·6H2O=X(X1=5%,X2=10%,X3=15%)]等试剂在集热式恒温加热磁力搅拌器剧烈搅拌下以2 min的间隔分别加入烧杯中后磁力搅拌20 min得到酒红色溶液。将此溶液转移至高压反应釜内,于170 ℃水热17 h,抽滤、洗涤数次,放置干燥箱中80 ℃下烘干。烘干后在马弗炉空气氛围中,以3 ℃/min从室温升至300 ℃焙烧200 min得到目标催化剂。
1.1.3 二次水热法制备Ce掺杂NiCo2O4催化剂
把1 mol/L Na2CO3溶液5 mL、无水乙醇12.5 mL、1 mol/L Co(NO3)2·6H2O溶液3.4 mL、氨水11.5 mL、1 mol/L Ni(NO3)2·6H2O溶液1.7 mL,在集热式恒温加热磁力搅拌器剧烈搅拌下以2 min的间隔分别加入,将前体溶液再搅拌20 min,将酒红色均匀溶液转移至高压反应釜内,高压反应釜在鼓风干燥箱中170 ℃水热17 h;洗涤抽滤,在干燥箱中80 ℃下烘干200 min,得到目标前体物。将其溶于34.1 mL无水乙醇中,按物质的量比Ce(NO3)3·6H2O:Co(NO3)2·6H2O=X(X=15%)加入 Ce(NO3)3·6H2O,使用搅拌器搅拌20 min;将均匀溶液转移至高压反应釜内,高压反应釜在鼓风干燥箱中170 ℃水热17 h;洗涤抽滤,在干燥箱中80 ℃下烘干 200 min,烘干后在马弗炉空气氛围中,以3 ℃/min升温至300 ℃焙烧200 min得到目标催化剂。
1.1.4 二次水热-二次焙烧法制备Ce掺杂NiCo2O4催化剂
先用1.1.1节方法制备得到一次水热法制备NiCo2O4目标前体物,把其溶于34.1 mL无水乙醇中,按物质的量比Ce(NO3)3·6H2O∶Co(NO3)2·6H2O=X(X=15%)加入Ce(NO3)3·6H2O,使用集热式恒温加热磁力搅拌器搅拌20 min;将均匀溶液转移至高压反应釜内,高压反应釜在鼓风干燥箱中 170 ℃ 水热17 h;洗涤抽滤,在干燥箱中 80 ℃ 下烘干200 min,烘干后在马弗炉空气氛围中,以3 ℃/min升温至300 ℃焙烧200 min得到目标催化剂。
1.2 催化剂表征方法
采用X射线衍射仪(D/Max-2500,Rigaku Corporation,日本)表征晶相并分析晶格参数,其中CuKα辐射(λ=0.154 06 nm)为40 kV和30 mA。对于所有催化剂,扫描角度为5°<2θ<80°,扫描速度为10 (°)/min;使用氮气物理吸附仪(ASIQM000100-6,Quantachrome,美国)氮吸附测量样品的比表面积测定(brunauer-emmett-teller,BET)、BJH理论方法测得孔容孔径分布。在N2吸附之前,将样品在473 K下脱气4 h以除去任何残留的水分或其他挥发物。压力范围p/p0=0.05~0.99;采用透射电子显微镜(transmission electron microscopy,TEM)(JEOL JEM 2100,日本)测量催化剂晶像及微观形貌。X射线荧光(X-ray fluorescence,XRF)光谱仪(AXIOS,荷兰) 基于莫斯莱定律,以特征X射线为基础进行元素的定性、定量分析。H2程序升温还原(H2temperature-programmed reduction,H2-TPR)装置(AutoChem2920,美国),用电子分析天平准确称取催化剂样品100 mg,将称量好的催化剂装入U型石英管反应器中,通入N2,以10 ℃/min的升温速率由室温升至500 ℃,在500 ℃下对催化剂进行预处理1 h,然后温度降至50 ℃,切换成还原气,以H2和N2的体积比为10∶90的混合气为还原气,待热导池稳定后,即可进行程序升温还原实验。其具体测试条件如下:还原气体流速为30 mL/min,升温速率为10 ℃/min,升温至900 ℃。
1.3 催化氧化降解实验
图1所示为催化活性评价仪器流程,利用固定床反应器在线催化评价装置(WFS-3015,天津先权工贸发展有限公司,中国)测试催化活性。催化剂填充粒度为40~60目,催化剂填充量为0.5 mL。通过高压恒流泵把液态甲苯抽到汽化室,180 ℃进行汽化后,利用合成空气吹扫甲苯至反应床层,反应后尾气进入气相色谱,测定甲苯浓度。进气空速为10 000 h-1,甲苯浓度为2 538×10-6。使用HT-FFAP毛细管柱和FID检测器,通过气相色谱仪(GC7900,上海天美科学仪器有限公司,中国)分析甲苯的入口和出口浓度,计算去除率。甲苯的去除率为

图1 仪器流程Fig.1 Instrument flowchart
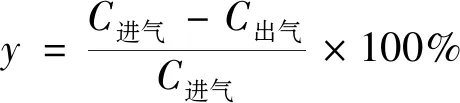
(1)
式(1)中:y为甲苯去除率,%;C进气为气相色谱进气口甲苯浓度;C出气为气相色谱进气口甲苯浓度。
2 结果与讨论
2.1 催化剂的催化活性
图2所示为尖晶石催化活性。使用温度T50、T90、T99(对应于甲苯转化率为50%、90%、99%)比较催化剂反应活性。总结图2结果制作表1。一次水热制备的15% Ce/NiCo2O4催化剂催化氧化甲苯显示出最优的活性(T50=249 ℃,T90=259 ℃,T90=267 ℃),经计算得出,在267 ℃时具有最高的甲苯转化速率[7.94 μmol/(gcats)]。比未掺杂铈的镍钴尖晶石具有更高的活性,且在267 ℃下催化氧化反应72 h后催化效率仅降低3.18%。即使反应过程中有水引入,催化活性也未受到很大的影响,表明此催化剂更能适应实际工业反应过程。
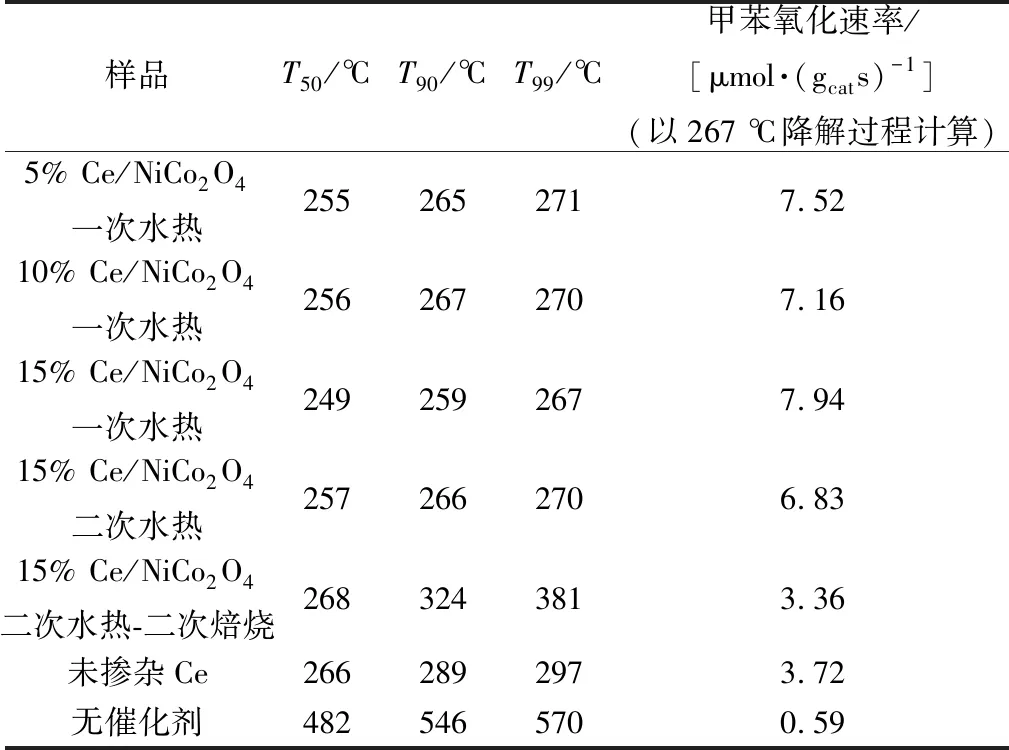
表1 T50、T90、T99对Ce/NiCo2O4的催化活性Table 1 Catalytic activities in terms of T50、T90 and T99 over Ce/NiCo2O4
2.2 催化剂的XRD分析
图3(a)所示为水热法制备15% Ce/NiCo2O4样品XRD图谱,可以看出水热法制备15% Ce/NiCo2O4样品均具有NiCo2O4尖晶石相(JCPDS-ICDD73-1702),在31°、36°、44°、59°、64°(2θ)处具有衍射峰,对应于(220)、(311)、(400)、(511)、(440)NiCo2O4晶面。水热法制备的15% Ce/NiCo2O4存在CeO2相(JCPDS-ICDD89-8436),在28°、47°(2θ)处有衍射峰,对应于(111)和(220)晶面。但一次水热法制备的15% Ce/NiCo2O4还存在CoO2相(JCPDS-ICDD70-3469),在23°(2θ)处有衍射峰,对应于(008)晶面。且一次水热制备的15% Ce/NiCo2O4显示最高的衍射峰强,结晶度最优,其中CoO2可能由于掺杂Ce后,部分Ce替换NiCo2O4中Co而形成的。15% Ce/NiCo2O4二次水热过程使样品结构坍塌,从而降低了尖晶石结晶度。而15% Ce/NiCo2O4二次水热二次焙烧过程,样品内部结构坍塌程度将会降低。如表2所示,由Scherrer公式计算水热法制备的15% Ce/NiCo2O4样品晶粒大小顺序为15% Ce/NiCo2O4一次水热法(2.40 nm)>15% Ce/NiCo2O4二次水热法(1.967 nm)>15% Ce/NiCo2O4二次水热-二次焙烧法(1.367 nm)。水热法制备的15% Ce/NiCo2O4样品晶胞尺寸相同均为 0.534 nm3,表明样品属于立方体晶系。
图3(b)所示为水热法制备xCe/NiCo2O4(x=5%、10%、15%)样品XRD图谱,可以看出水热法制备xCe/NiCo2O4(x=5%、10%、15%)样品均具有NiCo2O4尖晶石相(JCPDS-ICDD73-1702),在31°、36°、38°、44°、59°、64°(2θ)处具有衍射峰,对应于(220)、(311)、(222)、(400)、(511)、(440)NiCo2O4晶面。当掺杂量为5% Ce/NiCo2O4时,存在少量的CoO2相(JCPDS-ICDD70-3469),在23°(2θ)处有衍射峰,对应于(008)晶面。随着掺杂量的增加,10%Ce/NiCo2O4时,CoO相衍射峰的峰强明显增加,表示CoO的量增加而CoO2相消失。当15%Ce/NiCo2O4时,CoO相衍射峰基本消失,出现了CeO2相衍射峰(JCPDS-ICDD89-8436),在28°和47°(2θ)处有衍射峰,对应于(111)和(220)晶面,表明掺杂已经处于饱和状态。且随着Ce掺杂量的增加,催化剂中CoO2含量也明显上升,有效提高了高价态金属含量,且通过对一次水热、二次水热以及二次水热二次焙烧条制备的镍钴尖晶石进行氢气程序升温还原发现,一次水热制备的材料其还原峰向低温偏移,表明一次水热制备的催化剂具有更好的氧化还原性质,有利于活性氧的产生,对于高效催化氧化降解甲苯具有重要作用。同时晶粒计算结果如表2所示,由Scherrer公式计算水热法制备xCe/NiCo2O4(x=5%、10%、15%)样品晶粒大小顺序为10% Ce/NiCo2O4(3.70 nm)>5% Ce/NiCo2O4(2.767 nm)>15% Ce/NiCo2O4(2.40 nm)。较小的晶粒有利于催化过程中和目标污染物更好地接触,进一步提升催化反应效率。水热法制备的xCe/NiCo2O4(x=5%,10%,15%)样品晶胞体积相同均为0.534 nm3,表明样品属于立方体晶系。

表2 xCe/NiCo2O4(x=5%,10%,15%)样品XRD及XRF数据Table 2 XRD and XRF data of xCe/NiCo2O4(x=5%,10%,15%)
2.3 催化剂的BET分析
图4所示为xCe/NiCo2O4(x=5%、10%、15%)为N2吸脱附曲线和孔径分布,可以看出xCe/NiCo2O4(x=5%,10%,15%)属于典型的介孔分布曲线,BJH理论方法测得主要孔径分布在4 nm左右,如表3所示,xCe/NiCo2O4(x=5%,10%,15%)孔容BJH理论方法测得主要集中在0.1 cm2/g。从N2吸脱附曲线可知,迟滞环曲线属于H3型迟滞环回归线,在高压力区域没有表现出任何吸附限制。其中一次水热15% Ce/NiCo2O4在高压区域和低压区域都没有表现出任何吸附限制,表明该催化剂孔道结构均匀有序,可以对N2优先进行多层吸附。

P代表气体真实压力;P0代表气体在测量温度下的饱和蒸汽压;P/P0代表相对压力图4 xCe/NiCo2O4(x=5%,10%,15%)比表面积和孔容-孔径分布Fig.4 N2 adsorption-desorption isotherms and pore size distribution of xCe/NiCo2O4(x=5%,10%,15%)
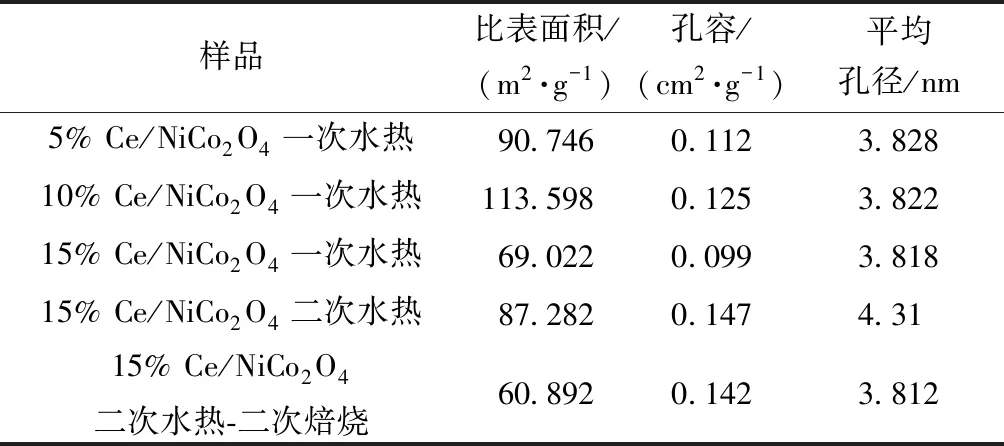
表3 材料氮气物理吸附结果Table 3 Textural properties of as-prepared materials
2.4 催化剂的TEM分析
图5所示为一次水热制备的15% Ce/NiCo2O4尖晶石催化剂TEM图。由图5(a)可以看出15% Ce/NiCo2O4形貌为棒状,从图5(b)、图5(c)可以看出棒状主要由颗粒堆积组成。图5(d)、图5(e)晶格尺寸分别为0.50 nm和0.28nm,对应的晶像分别为(111)和(220)。图5(f)为15%Ce/NiCo2O4衍射图。表明通过一次水热法,掺杂Ce可以得到形貌小颗粒堆积而成的棒状。

图5 15% Ce/NiCo2O4一次水热的TEM图和衍射图Fig.5 TEM and diffraction pattern of 15% Ce/NiCo2O4 after hydrothermal reaction
2.5 催化剂的催化氧化机理
如图6所示,采用一次水热掺杂Ce到NiCo2O4中制备尖晶石催化剂,随着掺杂量的增加,NiCo2O4中Ce达到饱和后以多余的CeO2形式存在。在氧化还原过程中,高价态的Co被还原,释放出活性氧原子即表面吸附氧,活性氧一部分参与反应,多余的则会储存在CeO2上。当甲苯达到催化剂表面时,表面活性氧起到主要降解作用,CeO2可通过自身氧化还原过程补充被逐渐消耗的活性氧,最终保持催化活性将甲苯有效分解为CO、H2O和CO2。图7所示为质谱数据,反应后未出现甲苯的质谱信号,表明已被完全降解。同时由于高价金属释放活性氧之后形成空穴,空气中氧会被空穴活化后重新形成活性氧,因此高价态金属含量越高,氧化还原性质越好,越有利于活性氧的生成。掺杂Ce主要有两方面作用:①制备具有较好的储-放氧特性的催化剂;②Ce的掺杂可提升催化剂中高价态金属含量,为活性氧的产生提供良好的条件。

图6 催化氧化机理Fig.6 Mechanism of catalytic oxidation

图7 质谱数据Fig.7 The data of GC-MS
3 结论
通过水热法将Ce掺杂到NiCo2O4中,并探究掺杂量对材料性能的影响,得出以下结论。掺杂Ce可以提高NiCo2O4中氧的生产速率。以甲苯为目标污染物,甲苯浓度为2 538×10-6,空速为10 000 h-1,催化剂体积为0.5 mL,一次水热15% Ce/NiCo2O4表现出最优的催化氧化性能,一次水热15% Ce/NiCo2O4的催化温度为T50=249 ℃、T90=259 ℃、T90=267 ℃;通过在267 ℃下连续催化氧化72 h后催化剂的催化氧化性能无显著变化,且引入5%水后对催化性能也没有产生任何影响,表明一次水热15% Ce/NiCo2O4催化剂具有良好的稳定性。
