Tumor necrosis family receptor superfamily member 9/tumor necrosis factor receptor-associated factor 1 pathway on hepatitis C viral persistence and natural history
2021-01-14JuliaPeAsensioEduardoSanzdeVillalobosJoaquMiquelJuanRamLarrubia
Julia Peña-Asensio, Eduardo Sanz-de-Villalobos, Joaquín Miquel, Juan Ramón Larrubia
Julia Peña-Asensio, Department of Systems Biology, Guadalajara University Hospital.University of Alcalá, Guadalajara E-19002, Guadalajara, Spain
Eduardo Sanz-de-Villalobos, Joaquín Miquel, Juan Ramón Larrubia, Translational Hepatology Unit, Guadalajara University Hospital, University of Alcalá, Guadalajara E-19002, Guadalajara, Spain
Abstract Hepatitis C virus (HCV) infection is an excellent immunological model for understanding the mechanisms developed by non-cytopathic viruses and tumors to evade the adaptative immune response.The antigen-specific cytotoxic T cell response is essential for keeping HCV under control, but during persistent infection, these cells become exhausted or even deleted.The exhaustion process is progressive and depends on the infection duration and level of antigenemia.During high antigenic load and long duration of infection, T cells become extremely exhausted and ultimately disappear due to apoptosis.The development of exhaustion involves the impairment of positive co-stimulation induced by regulatory cytokines, such as transforming growth factor beta 1.This cytokine downregulates tumor necrosis factor receptor (TNFR)-associated factor 1 (TRAF1), the signal transducer of the T cell co-stimulatory molecule TNFR superfamily member 9 (known as 4-1BB).This impairment correlates with the low reactivity of T cells and an exhaustion phenotype.Treatment with interleukin-7 in vitro restores TRAF1 expression and rescues T cell effector function.The process of TRAF1 loss and its in vitro recovery is hierarchical, and more affected by severe disease progression.In conclusion, TRAF1 dynamics on T cells define a new pathogenic model that describes some aspects of the natural history of HCV, and sheds light on novel immunotherapy strategies for chronic viral infections and cancer.
Key Words: Hepatitis C virus; Tumor necrosis factor receptor-associated factor 1; CD8; Exhaustion; Tumor necrosis family receptor superfamily member 9; Chronic hepatitis
INTRODUCTION
Hepatitis C virus (HCV) evolution is heterogenous as a result of the particular interplay between the virus and the immune system[1].The outcome of the fight between host and pathogen depends on the balance of the host-microbe interaction, which causes varying degrees of progressive liver damage[2-5].The fine-tuning of this equilibrium can induce either rapid or slow disease progression, which depends on the degree of impairment of the adaptive immune system[6].During persistent noncytopathic viral infection, the antigen (Ag)-specific T cell response is exhausted and unable to clear infection despite achieving partial viral control[7,8].The correct activation of this response relies on the interaction with Ag-presenting cells (commonly known as APCs) in the proper cytokine environment with the right costimulation[1,9,10].Non-cytopathic viruses manipulate T cell co-stimulation for their own benefit, favoring the induction of negative co-stimulatory receptors and inhibiting positive co-stimulatory pathways[11-13].Tumor necrosis factor receptor (TNFR) superfamily member 9 (4-1BB) is a TNFR-associated factor 1 (TRAF1)-binding checkpoint molecule that is normally absent from resting cells but is induced by T cell receptor (TCR) signaling[14].It is a positive activator of the T cell response, which is key during viral infection and cancer.TRAF1 is the major signal transducer after 4-1BB triggering[15], and its downregulation on T cells is used by pathogens as a mechanism to evade specific adaptive immune responses[2,16].
In this review, we present an update on the current knowledge of the role of the 4-1BB/TRAF1 pathway in the outcome of HCV infection, and how it can be manipulated to overcome T cell exhaustion.Although this immunotherapeutic strategy is no longer needed in the era of direct acting anti-viral (commonly referred to as DAA) medications[17,18], lessons obtained from this persistent infection model can be extrapolated to other viral infections, such as hepatitis B virus (known as HBV) and human immunodeficiency virus (HIV), or cancer.
ROLE OF T CELLS IN THE NATURAL HISTORY OF HCV
HCV is a highly variable, positive-sense, single-stranded hepatotropic non-cytopathic RNA virus of the familyFlaviviridae[19,20], with parenteral, vertical, and sexual transmission capacities[21].HCV induces progressive liver damage that can lead to chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma[3,22].About one-third of patients spontaneously clear the virus but in the remaining two-thirds, the infection persists unless an anti-viral treatment is administered[5].Currently, the infection is easily controlled by using DAA drugs[17].Nevertheless, it is still possible to learn from HCV about the host-pathogen interaction in chronic viral diseases, which can be applied to other chronic viral infections and cancer.
During the natural history of untreated, persistent HCV infection, there are three different progression groups: Slow, mild, and rapid fibrosers.Slow fibrosers do not develop significant fibrosis during their life, whereas rapid fibrosers can progress to cirrhosis, portal hypertension, or hepatocellular carcinoma in as quickly as 10-20 years after primoinfection[23].Host factors such as sex, age of infection, alcohol consumption, co-infection with HIV or HBV, steatosis, and insulin resistance[23,24], as well as the quality of the adaptive immune response[1], are involved in the different evolution patterns of HCV.HCV-specific cytotoxic T cells play a central role in controlling HCV infection[25,26].During persistent HCV infection, however, the cytotoxic T cell response becomes dysfunctional, with cells presenting markers of exhaustion and apoptosis[27-30].Nevertheless, these HCV-specific CD8 T cells can still partially control viral replication[31].
Interestingly, it is not HCV-specific CD8 T cells but other inflammatory cells recruited to the infected liver that are ultimately responsible for persistent liver damage[32,33](Figure 1).Therefore, long-lasting infection linked to a weak CD8-specific T cell response can induce permanent non-specific inflammatory infiltrates that can promote the rapid progression of liver fibrosis[33,34].In fact, a high level of prolonged antigenemia induces a hierarchical loss of effector functions and ultimate apoptosis of T cells[35].During persistent HCV infection, the level of specific T cell impairment positively correlates with the speed of liver fibrosis progression.These data suggest that stronger T cell exhaustion may facilitate rapid fibrosis progression.In support, rapid fibrosers with long-lasting infection lack detectable peripheral HCV-specific cytotoxic T cells, which although exhausted, are present in slow fibrosers and shortterm disease[2].Consequently, it may be possible to restore specific T cell responses to improve viral control, and in addition, to prevent liver damage by reducing proinflammatory chemokines and cytokines secreted in the infected liver.
During chronic hepatitis C, some pro-fibrogenic and immunoregulatory cytokines, such as transforming growth factor beta 1 (TGF-β1) are increased.In vitroanalysis has shown that after Ag encounter, HCV-specific CD8 T cells secrete TFG-b1, which is linked to effector dysfunction and can be rescued by anti-TGF-β1 blocking antibodies[36].Moreover, HCV itself is able to induce liver cells to express TGF-β1, and the number of TGF-β1-secreting regulatory T cells is also enhanced during chronic hepatitis C infection[37,38].Among its immunoregulatory properties, TGF-β1 has been linked with the negative modulation of the positive co-stimulatory checkpoint 4-1BB/TRAF1 in some chronic viral infections, such as those by HIV, HCV, and lymphocoriomeningitis virus[2,16].
In the next sections of this review, this specific pathogenic axis will be discussed in detail.
4-1BB/TRAF1 PATHWAY
4-1BB, also called CD137, is a co-stimulatory checkpoint that is predominantly expressed on activated CD8 T cells and natural killer cells[39], and in lower levels on CD4 T cells, dendritic cells, granulocytes, and mast cells[40].It binds to 4-1BB-ligand (4-1BBL, CD137L, or L/TNFR9), which is present on such APCs as activated B cells, dendritic cells, and macrophages[41]; the 4-1BB/TRAF1 pathway is shown in Figure 2.4-1BBL trimer has a three-bladed propeller structure and binds to three 4-1BB receptor monomers[42].4-1BB translocates to the membrane after Ag encounter on CD8+T cells[43], recruiting the TRAF family members TRAF1, 2, and 3[44].Signaling through the 4-1BB receptor depends on the association with TRAF1 and 2 molecules, as evidence shows that the lack of any of them blocks 4-1BB/4-1BBL downstream transduction[16,45].
TRAF 1, 2, and 3 can form heterodimers and interact with adaptor proteins (i.e., ubiquitin ligases, proteases, kinases), creating a three-dimensional structure complex where enzymatic processes can be carried out[46].TRAF1 differs from the other members of its family, as it lacks the N-terminal RING finger domain, which prevents it from acting as an E3 ubiquitin ligase.However, TRAF1 acts as a bridge between a wide range of adaptor proteins, regulating their activity[47]and interacting with several TNFR members, prompting their stimulation or inhibition.TRAF1 has a role in T cell activation through the canonical nuclear factor-kappa B (NF-κB) pathway and an alternate pathway.These two different mechanisms of action regulate the physiology of T cells.In the canonical pathway, TRAF1 is inducible after cell activation through NF-κB[48], and is present in a restricted group of cells in which activated lymphocytes are included[49].TRAF1 regulates survival signals mediated by TRAF2, modulating their ability to mediate sustained activation of NF-κB and c-Jun N-terminal kinase[50].Specifically, TRAF1 is implicated in extracellular signal-regulated kinase (ERK) activation mediated by leukocyte-specific protein 1[51].

Figure 1 Theoretical model of liver damage during chronic viral hepatitis due to non-specific inflammatory infiltrate.
ERK phosphorylates Bim, eliciting its elimination by the proteasome and abrogating its anti-apoptotic effects[52].The formation of two heterotrimers TRAF1:TRAF2 results in the recruitment of cellular inhibitor of apoptosis protein (cIAP) as well as the interaction with other adaptor proteins and protein kinases, which leads to activation of the NF-κB pathway[53].TRAF2 can also dimerize to activate E3 ubiquitin ligases through their RING finger domains.Evidence indicates that the interactions among different TRAFs heterodimers allow them to adopt an octagonal superstructure where many 4-1BB/4-1BBL act simultaneously.This structure has been called the 4-1BB signalosome and could provide a model to design novel 4-1BB analogues as immunotherapeutic strategy[46].Downstream signaling leads to the phosphorylation of inhibitor of kappa B kinase subunit β and subsequent activation of canonical NF-κB[54], ERK1/2[55],and p38 mitogen-activated protein kinase[56].Collectively, this 4-1BBdependent modulation results in CD8 T cell proliferation and survival.
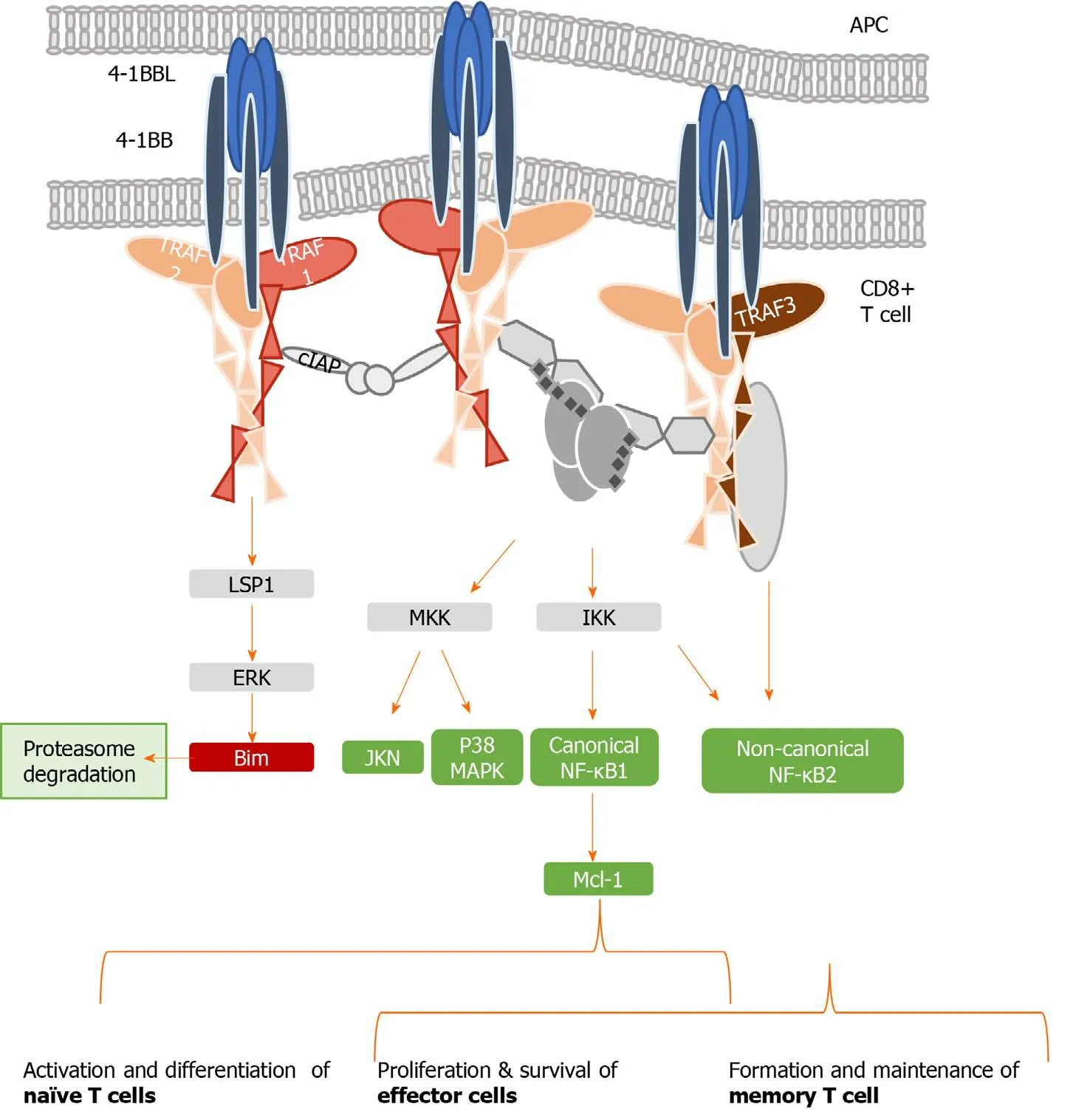
Figure 2 Tumor necrosis family receptor superfamily member 9/tumor necrosis factor receptor-associated factor 1 signaling complex.
When TNFR signaling is active, TRAF1 also engages the non-canonical NF-κB pathway by degrading TRAF3[54,57,58].Initiation of the non-canonical NF-κB pathway is delayed with respect to the canonical one, which may play a role in T cell activation and memory differentiation[56].Thus, in contrast to the rapid and transient activation of the canonical NF-κB pathway, activation of the non-canonical NF-κB pathway is characteristically slow and persistent.On the other hand, TRAF1 also regulates the canonical pathway by preventing TRAF2 degradation or enhancing cIAP recruitment, degrading NF-κB-inducing kinase, which is necessary for activation of the alternate NF-κB pathway[14,58].Therefore, TRAF1 is a key transducer involved in initial T cell activation and proliferation by the canonical NF-κB pathway, but also in the generation of the memory and effector pool in a delayed manner through the noncanonical NF-κB pathway[54,56].
Figure 2 summarizes the different pathways involved in 4-1BB signaling.
4-1BB/TRAF1 AND SPECIFIC CYTOTOXIC T CELL RESPONSE
Cytotoxic T cells carry out an essential task in non-cytopathic virus control[59,60].This population is able to recognize infected cells and clear the virus by cytopathic and non-cytopathic mechanisms.Follow-up of healthcare workers after accidental needlestick HCV exposure showed that in those who naturally controlled the virus, HCV-specific CD8 T cells initially destroyed some hepatocytes but later removed the virus by releasing interferon-g[60].These immune cells become activated by the combination of three different signals.First of all, the interaction between the APC and the TCR is necessary[61].Thereafter, the interleukin (IL)-2 receptor is upregulated and its subsequent activation promotes T cell proliferation[62].These two signals must be combined with the activation of early and late positive co-stimulatory checkpoints.Early positive co-stimulatory CD27 and CD28 counteract the inhibitory effects of negative checkpoints such as programmed cell death protein-1 (PD-1)[63-65].Late positive co-stimulatory molecules such as 4-1BB play an important role in boosting the T cell response and inducing memory generation[14,66].
The 4-1BB/TRAF1 pathway promotes T cell memory formation[67]and survival[55,68]but also regulates effector T cell trafficking into the infected organ[69].The triggering of this pathway can also improve T cell effector function by mitochondrial morphological and functional reprogramming[12,70,71].Noteworthy, 4-1BB co-stimulation activates glucose and fatty acid metabolism to enhance CD8 T cell reactivity[72].As noted above, the role of 4-1BB in T cell survival is mainly mediatedviaERK by the downregulation of the pro-apoptotic protein Bim[55,73,74].Thus, pharmacological intervention of this pathway can improve the T cell response by increasing survival and reactivity.
Tumors and persistent viral infections counter positive co-stimulation by early induction of negative checkpoints and inhibition of the positive checkpoints[7].During non-cytopathic persistent viral infections, specific CD8 T cells are characterized by the expression of negative co-stimulatory molecules such as PD-1, T cell immunoglobulin and mucin-domain containing-3, and cytotoxic T-lymphocyte protein 4[11,27,75].In addition, these viruses can impair downstream signaling of 4-1BB by causing the loss of its signal transducer TRAF1[16], which explains why positive immunotherapeutic modulation of 4-1BB has failed to boost the virus-specific CD8 T cell response[76].During chronic lymphocytic choriomeningitis virus infection in mice, TRAF1 Loss on specific CD8 T cells is caused by TGF-β1-induced TRAF1 degradation, and this effect can be counter-regulated by common-g chain receptor cytokines, such as IL-7[16].
Interestingly, similar data have been reported for some human infections.Particularly, in chronic progressors during HIV infection, TRAF1 expression is lower than in elite controllers[16].T cells from those elite controllers are more active in controlling HIV-infected cells and the process is correlated with TRAF1-mediated Bim downregulation.Indeed, the T cell response during HCV infection shares many features with HIV, and consequently, TRAF1 signaling could also be involved in HCVspecific T cell exhaustion, as will be discussed in the next section.
TRAF1 INVOLVEMENT IN HCV T CELL EXHAUSTION
Exhausted HCV-specific cytotoxic T cells are characterized by the high expression of negative checkpoint proteins, such as PD-1, and low expression of the IL-7 receptor CD127[27](Figure 3).Lack of CD127 makes these cells less sensitive to the pro-survival cytokine IL-7, which stabilizes the anti-apoptotic protein myeloid leukemia cell differentiation protein (Mcl-1)[28](Figure 3).IL-7/IL-7R signaling positively regulates Mcl-1viasignal transducer and activator of transcription 5[77]but also increases TRAF1 level[16](Figure 4).As previously stated, 4-1BB/TRAF1 also counters BimviaERK signaling[55](Figure 4).Moreover, during persistent HCV infection, TGF-β1 Level is increased, and this cytokine downregulates TRAF1 expression on T cells.Hence, during HCV infection, the combination of low IL-7 sensitivity linked to the higher TGF-β1 Level could be the “perfect storm” to desensitize 4-1BB signalingviaTRAF1 Loss.This suggests that, as in HIV infection[16], the loss of TRAF1 in HCV-specific CD8 T cells during chronic hepatitis C is central to the aforementioned imbalance between Bim and Mcl-1[28](Figures 2 and 3).Therefore, HCV-specific T cells could be poorly reactive and prone to apoptosis due to the lack of signaling by IL-7 and 4-1BB.
TGF-β1 Levels are increased during persistent HCV infection[2,36,37]and there is low IL-7 receptor expression on T cells.TRAF1 is positively and negatively regulated by IL-7 and TGF-β1, respectively[16].With this in mind, we hypothesize that high TGF-β1 Level during HCV infection could downregulate TRAF1, impairing 4-1BB signaling and upregulating Bim.Furthermore, low CD127 expression on HCV-specific CD8 T cells would also reduce Mcl-1 Levels.The combination of low Mcl-1 and high Bim levels would synergize to negatively affect T cell proliferation, cytotoxicity, and survival (Figure 4).
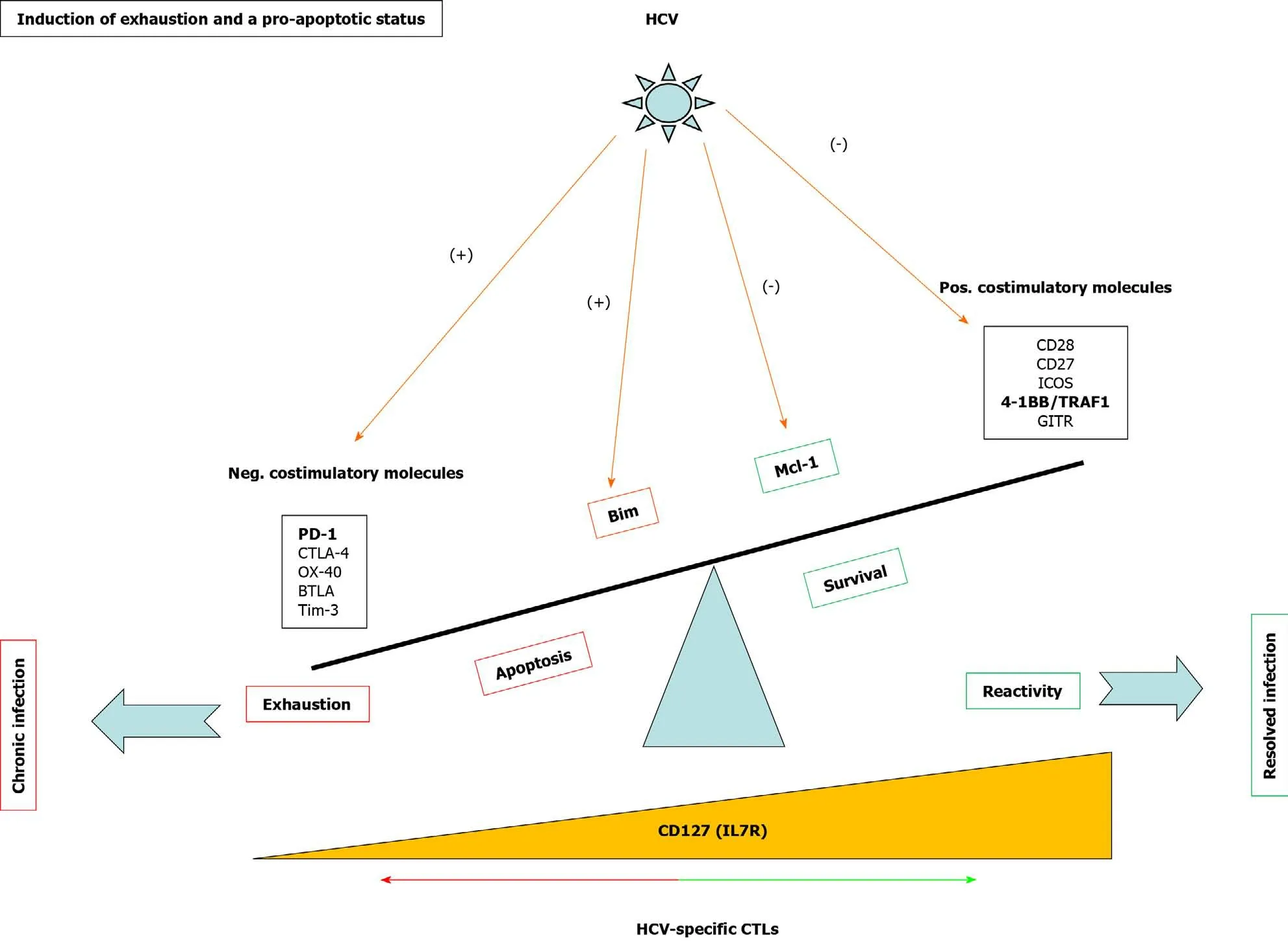
Figure 3 Mechanisms involved in T cell exhaustion and apoptosis during persistent hepatitis C virus infection.
To test this hypothesis, our group detected TRAF1 expression directlyex vivoon HCV-specific CD8 T cells from chronically-infected and treated patients.As was expected, those individuals with persistent viral replication had lower TRAF1 expression than HCV controllers[2].Moreover, TRAF1 expression was inversely correlated with the exhausted and pro-apoptotic phenotypes and directly correlated with T cell reactivity.Low TRAF1 expressing T cells were PD-1high, Mcl-1low, and CD127low, and did not expand after Ag encounter.Analysis of the supernatants of Agspecific T cell cultures showed that those cases with less proliferative potential had higher levels of TGF-β1.Moreover, a negative correlation was also observed between serum TGF-β1 Level and TRAF1 expression on Ag-specific CD8 T cells.Furthermore, TGF-β1in vitrotreatment of HCV-specific CD8 T cells from resolvers induced TRAF1 downregulation, and this effect was counteracted by IL-7 treatment.Although the CD127 expression level is low in the effector progeny subset, the low frequency progenitor pool still maintains this receptor, and it is this population that is suitable for immunotherapy[78,79].Moreover, IL-7 at a therapeutic dose can antagonize multiple cellular and molecular networks[80].These data suggest that during persistent HCV infection, TGF-β1 downregulates TRAF1 in T cells, which can be reversed byex vivoIL-7 treatment.

Figure 4 Tumor necrosis factor receptor-associated factor 1 pathways involved in T cell survival.
Consequently, we developed an IL-7 and 4-1BBL combination treatment to improve T cell reactivity; IL-7-dependent upregulation of TRAF1 restored 4-1BB signaling to fully enable the agonist actions of 4-1BBL over 4-1BB.We observed a hierarchical response that was dependent on the stage of HCV infection; only cases with less severe fibrosis and lower evolution responded favorably to the 4-1BBL/IL-7 combination[2].We speculated that cases with worse progression probably had higher burden of exhausted T cells with increased PD-1 expression, leading us to add anti-PD-L1 treatment to the IL-7/4-1BBL combination[81].After the combined treatment, we were able to restore two other groups of cases: Those with low fibrosis progression but longterm infection, and those with rapid-progression and short-lasting disease.Unfortunately, those cases with less favorable factors, specifically rapid fibrosis progressors with long-term infection, were not responsive to the treatment[2].This may have been due to the loss of these T cell populations from apoptosis (Figure 5).
CONCLUSION
The HCV-specific T cell response impacts infection outcomes.Mid-slow fibrosis progressors have less exhausted T cells, but the length of infection also influences the impairment of the T cell response.Worse T cell reactivity is observed the longer the infection lasts, and the faster liver fibrosis takes place.T cell response impairment is mediated by an exhausted and pro-apoptotic status that is characterized by the upregulated expression of negative checkpoints and the inhibition of positive costimulatory molecules.Among the latter is 4-1BB signalingviaits effector TRAF1.This pathway regulates downstream BimviaERK and is involved in T cell activation and survival.TRAF1 is induced by IL-7 and downregulated by TGF-β1.During persistent HCV infection, TGF-β1 Level is increased and can contribute to T cell exhaustion by TRAF1 loss.Depending on the stage of the infection, IL-7ex vivotreatment can restore TRAF1 expression and T cell reactivity (Figure 5).
4-1BB/TRAF1 has a pathogenic role in chronic HCV infection that describes a new mechanism of T cell exhaustion and explains different infection outcomes.Modulation of 4-1BB/TRAF1 can be useful as an immunotherapeutic strategy in chronic viral infections and cancer.

Figure 5 Tumor necrosis factor receptor-associated factor 1-related pathogenic mechanism involved in T cell exhaustion and liver fibrosis progression during persistent hepatitis C virus infection.
杂志排行

World Journal of Hepatology的其它文章
- Neoadjuvant treatment strategies for intrahepatic cholangiocarcinoma
- Metabolic syndrome and liver disease in the era of bariatric surgery: What you need to know!
- Combined liver-kidney transplantation for rare diseases
- Hepatocellular carcinoma Liver Imaging Reporting and Data Systems treatment response assessment: Lessons learned and future directions
- Apatinib as an alternative therapy for advanced hepatocellular carcinoma
- Hepatitis B virus detected in paper currencies in a densely populated city of India: A plausible source of horizontal transmission?