奎宁手性催化合成苯并噻唑氨基酸酯反应机理研究
2021-01-13谢承卫高弦
谢承卫 高弦



Abstract: The dissymmetric Mannich reaction of benzothiazol-β-amino esters is of great importance for exploring effective enantioisomer with good bioactivity. The mechanism for Mannich reaction of benzothiazol-β-amino esters catalyzed by simple chiral quinine organocatalyst was investigated through a combination of experiment with theoretical approaches (DFT). With solvent effect taken into consideration, transition states TS (S or R) were confirmed with potent strategy of hybrid density functional M06-2X at the level of 6-311G(d, p) basis set. The key feature of dual activation mechanism lies in the formation of one hydrogen bond O (12)—H (25)—N (57) related to quinine hydroxyl (Cat) and benzothiazol imines (EI) N (57) and the other hydrogen bond N(1)—H(90)—O(79) formation related to tertiary amine of quinine (Cat), by which diethyl malonate is able to be activated into enolic Nu simultaneously. The result obtained through energetic calculation was identified further by IR vibrating frequency to convince of transition state attained to be accurate. As a comparison of (R)TS pathway with (S)TS pathway in potential energy profile, it enables to elaborate that (S) TS pathway executes to afford unique enantioisomer (S). At the same time, one of reacting factors was optimized for increasing enantio-and distereoselectivity using DFT calculations, that is reacting temperature. The lower the temperature is going on, the more the enantio-and distereoselectivity are upgraded.
Key words: chiral quinine; benzothiazol-β-amino esters; DFT; transition state; mechanism
中圖分类号:O643.3 文献标志码:A
基金项目:贵州省自然科学基金资助项目([2017]1028)
Dissymmetric organocatalysis has been exploited as a potent approach for the various synthesis of chiral compounds. Design, application and mechanistic investigation on new activation modes (such as enamine, iminium, and hydrogen bonding catalysis) have dramatically advanced the development in this field[1-3]. In 2010, the highly enantioselective organocatalytic Friedel-Crafts-type addition of indole to isatin catalyzed by cinchona alkaloid derivatives was reported by CHAUHAN and co-worker[2]. This group also had proposed rational mechanism relying on hydrogen bonding interaction. In the following year, ALESSIO and ALESSANDRA[3] used unmodified cinchona alkaloid as organocatalyst to asymmetric conjugate addition of diarylphosphane oxides to chalcones with good yield and up to 89% ee. It’s worthy to note that, since entering modern era, computation science has been developed fast, computational calculation has been serving as a reliable strategy for analyzing reacting mechanism instead of traditional theoretic inference. To the best of our knowledge, mechanism plays an important role in dissymmetric transformation. Mechanism as theoretic guidance explains detail process for reactions in functionality. In 2012, ZHU and co-workers[1] investigated on dual activation mechanism for the direct vinylogous Michael reaction of α, β-unsaturated γ-butyrolactam and chalcone catalyzed by the bifunctional cinchona alkaloid thiourea orgnaocatalysts by NMR and DFT calculations, which stated N-HB bond of thiourea moiety activating EI was weaker than both of N-H bond of protonated amine and N-HA bond of thiourea moiety activating Nu to gain agreeable results via theory calculation. In 2008, BUSYGIN and co-workers[4] explored some cinchona alkaloid o-ethers conformation by means of combining methods NMR, DFT and X-ray.
Benzothiazol-β-amino esters, acted as β-amino acids derivatives, perform excellent bioactivity in antibacterial and antivirus[5-7]. To gain highly and purely optical single enantioisomer of benzothiazol-β-amino esters with simple organocatalyst, recently, the development for an classic Mannich reaction of diethyl malonate (Nu) with benzothiazol imine (EI) in the presence of cinchona alkaloid derivatives as organocatalyst, including chiral quinine organocatalyst, cinchona alkaloid thiourea organocatalyst was reported by LI and BAI[8-9] with good enatioselectivity of up to 81%~99% ee. In this paper, we carry out mechanistic investigation on representative example (Scheme 1) of this reaction via experiment and theoretic approaches (DFT). Through analyzing potential energy profile of transition state, the most stable transition state is confirmed to get preferable pathway for this reaction. To obtain more reliability for revealing truly reacting process, we originally applied the newly method M06-2X to the calculation of potential energy, bond-length, and hydrogen-bonding intensity at the level of 6-311G(d, p) basis set to confirm the most transition state in asymmetric organocatalysis.
1 Materials and methods
1.1 Experimental Section
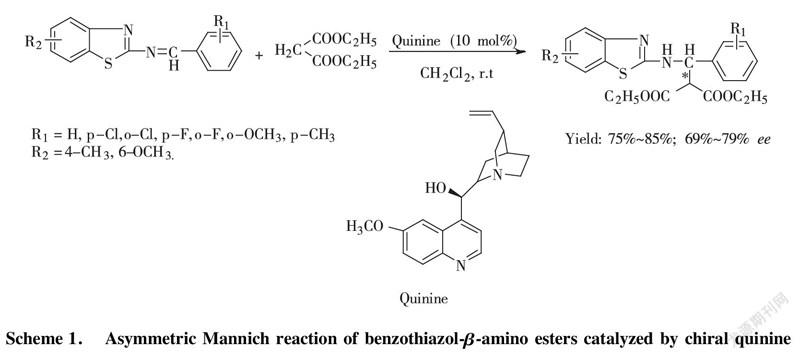
Scheme 1. Asymmetric Mannich reaction of benzothiazol-β-amino esters catalyzed by chiral quinine
To solute the benzothiazol imine (1.0 mmol) in DCM (3.0 mL) chiral quinine catalyst, loading equivalent 10 mol% was added to stir at room temperature. After 5 min, diethyl malonate (1.2 mmol) was put into reacting system dropwise and keeping stirring for 72~96 h. After working up, target compound was separated and purified by column chromatography with hexane/ethyl acetate=5/1 to 3/1 as elute.
1.2 Computational Methods
In depth to understanding the real reacting process, all geometry structures of Cat, two substrates(Nu and EI)were first optimized completely with the moderate 6-311G(d, p) basis set in the gas phase. Quantum chemical calculations were exhibited by means of the hybrid density functional approach M06-2X in the Gaussian 09 program suite. The reacting transition states were confirmed by calculating its vibrating frequency at the same level of 6-311G(d,p) basis set as implemented[10-11]. As a result of calculating inaccuracy for hydrogen bonding interaction of catalyst with substrates by employing hybrid density functional method B3LYP[12-16], a method M06-2X was utilized further to improve calculating accuracy for hydrogen bonding interaction in variable molecular system with moderate 6-311G(d,p) basis set.With effect of DCM as solvent,CPCM polarization continuum model[17] was used. Afterward, frequency calculation were performed at the same level of 6-311G(d, p) basis set as the geometry optimizations to confirm the minimum and transition state (TS) structure (zero and only one imaginary frequency respectively), and then to ascertain activating combination of catalyst with substrates that is involved in calculation for all saddles placed on potential energy surface in frequency. Intrinsic reaction coordinates (IRC)[18-19] were employed for monitoring progress of reaction, verifying formation of transition state, so that all saddle points were connected to reactant and product at the same level of basis set. Finally, the energy data were derived from moderate approach M06-2X/6-311G(d,p), including the zero-point energy (ZPE) correction from frequency calculation and the solvent CH2Cl2 effect(CPCM). All of structure images were generated using Gaussian 09 program suite[20]. All of the bond lengths are in angstroms (A), and the energies are in kJ/mol.
2 Results and Discussion
2.1 Molecular Structures of Substrates
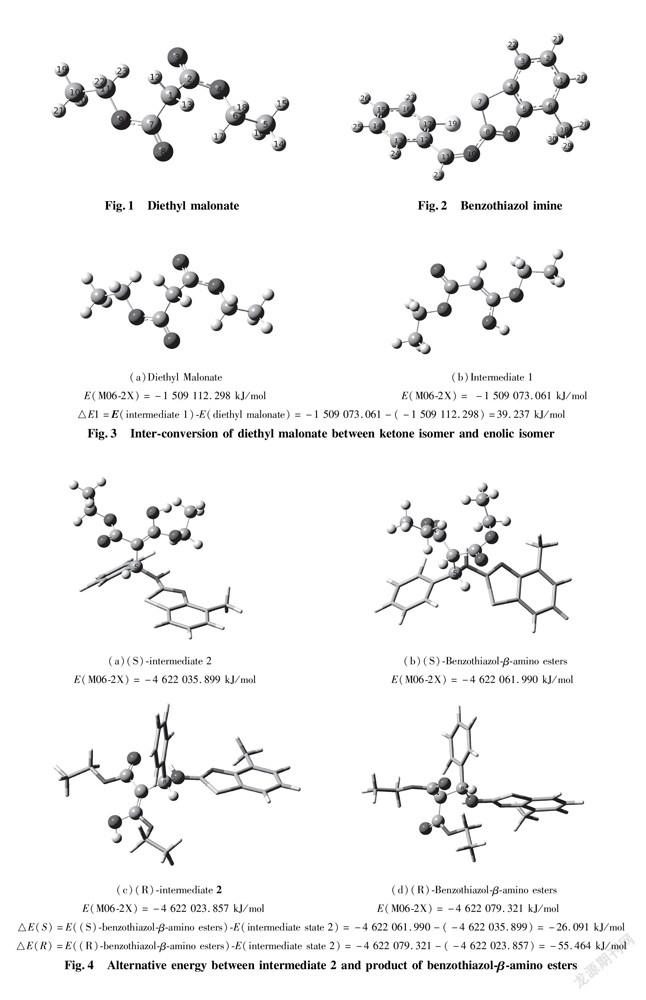
The formation of hydrogen bonds among Cat, Nu and EI in distereoselectivity is regarded as a predominant feature. Once the combination of catalyst with substrates through hydrogen bonding interaction takes place, spatial orientation of collision between Nu and EI has to proceed at one direction to increase distereoselectivity in presence of chiral quinine organocatalyst. Because formation of hydrogen bonds between substrates and catalyst requires strong electronegative atoms as participants, it is a core for dissymmetric transformation. We first carried out optimization to the two substrates structure of nucleophiles and eletrophiles to afford their most stable conformations. One of substrates, diethyl malonate (Fig.1) as nucleophile containing four sorts of oxygen atoms O(3),O(4),O(8),O(9) are to be possible active sites; the other is benzothiazol imine(Fig.2)as electrophile having four strong electronegative atoms S(7),N(9),N(10),F(19) are also to be possible active sites. On one hand, as described in Fig.3, in the view of energy, the energy barrier of isomer ketone is almost equal to that of the other isomer enol in process of inter-conversion of diethyl malonate(△E1=39.237 kJ/mol), so it is easy to make conversion of ketone isomer of diethyl malonate into corresponding enolic isomer as intermediate state 1(int.1). The enolic isomer is able to donate an active proton, considered as a donor in the formation of hydrogen bonds. On the other hand, the structure frame of benzothiazol imine performs the most stable configuration through optimization to illustrate that it is not planar molecular, even though executing p orbits conjugation, as depicted in Fig.2.
In addition, before benzothiazol-β-amino esters obtained, reacting substrate benzothiazol imines and diethyl malonate are allowed to afford intermediate state 2(int. 2), and then to make inter-conversion between ketone isomer and enolic isomer. The result is determined by △E(S)(-26.091 kJ/mol) or △E(R)(-55.464 kJ/mol)as shown in Fig. 4.
2.2 Quinine Reaction Coordination
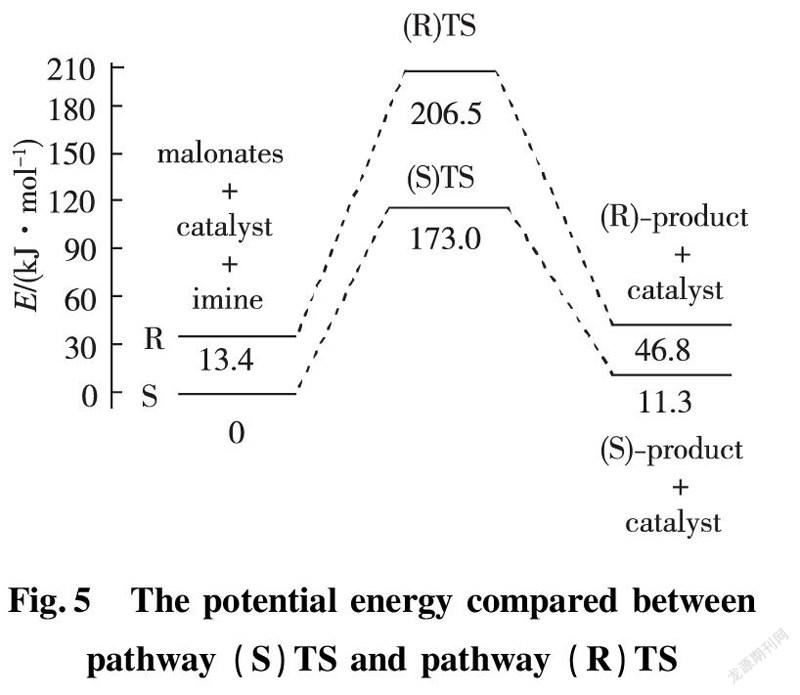
In application of chiral quinine as organocatalyst to dissymmetric Mannich reaction of benzothiazol-β-amino esters, there are two possibilities exhibited in enantio-and distereoselcetivity. In order to directly put insight into asymmetric transformation, the calculating potential energy profiles of two pathways (S)TS and (R)TS are compared to discover more stable transition state. All energies are relevant with the total energy of the most stable conformer of Cat, EI and enolic Nu. The total energy of catalyst and reactant is 13.4 kJ/mol at the outset of reaction. The energy barrier for transition state (S) TS is 173.0 kJ/mol, lower obviously than that of (R) TS with 193.1 kJ/mol, which explicitly indicates pathway (S)TS is predominant procedure as shown in Fig.5.
Since the active energies differ little between them, increasing the temperature would result in an accelerated rate-determining step of the pathway (R) TS, thereby reducing the distereoselectivity.Therefore, controlling reacting temperature is one of the vital factors for high distereoselectivity. The factor is completely in agreement with experimental results under reacting temperature lower than 25 ℃, product was afforded with S configuration, arriving at 79% ee.
2.3 Violating Diagnosis and Confirmation of Transition States
2.3.1 Analysis of Imaginary Vibrating Frequency
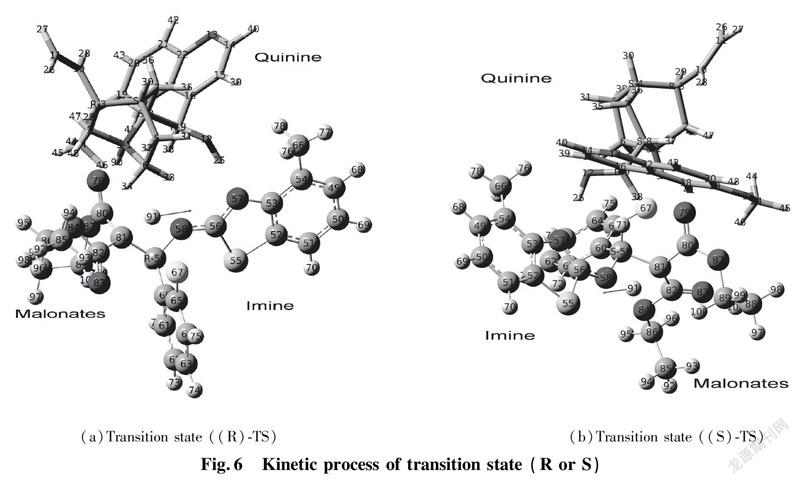
In this section, we discussed about vibrating frequency of transition states to confirm active sites. We employed the DFT calculations to explore active sites among catalyst, EI and enolic Nu. According to the results of calculations, the values of vibrating frequency of reactants, intermediates (int.1 and int.2) and products are positive, which demonstrate that they are in certain area of lowest point. As for calculations for transition state (R) TS and (S) TS, both of them are equipped with unique imaginary frequency respectively. That frequency derives from H(91) atom of diethyl malonate vibration in the area between C(81) of diethyl malonate and N(58)atom of benzothiazol imine as shown in Fig.6. Consequently, by diagnosis for intensity of imaginary frequency and vibrating positions of corresponding atoms, we find out the most stable transition state. (a)Transition state ((R)-TS) (b)Transition state ((S)-TS)
2.3.2 IR Vibrating Frequency
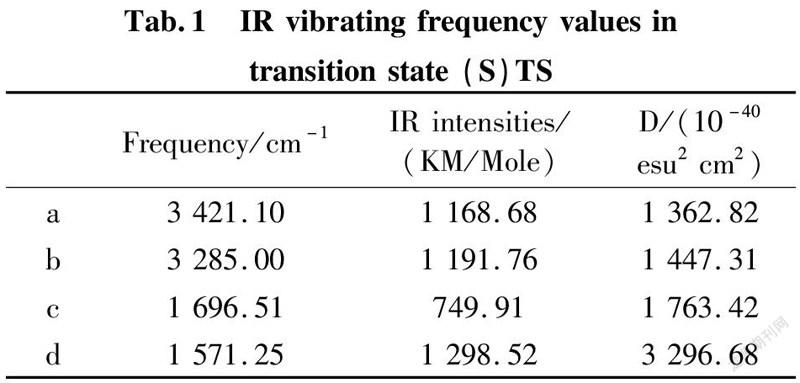
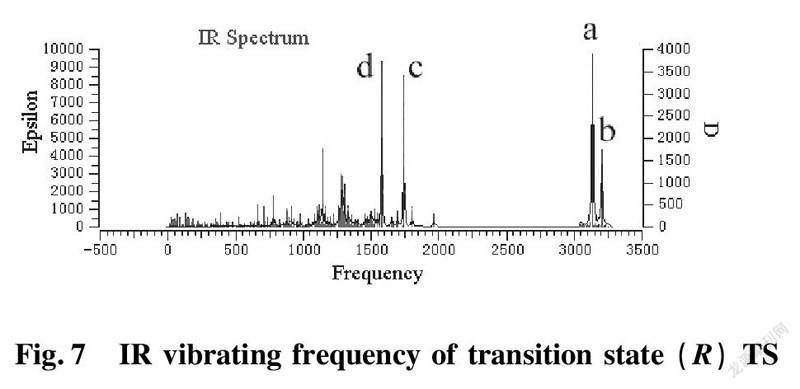
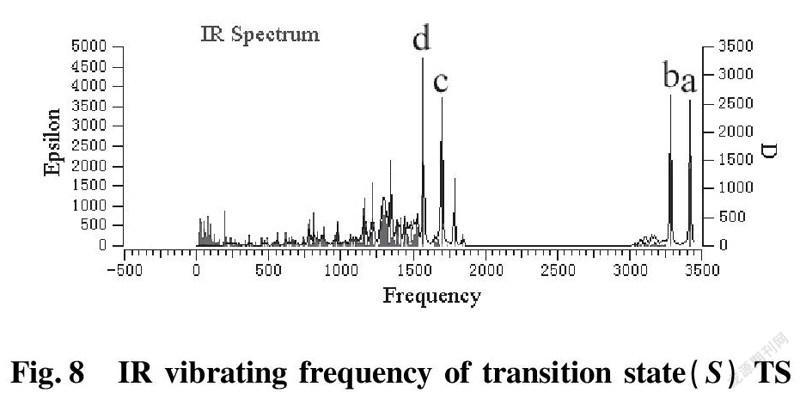
To further validate reliability of transition state obtained, we conducted the DFT calculation for transition states (R)TS and (S)TS with moderate approach IR vibrating frequency, as shown in Fig.7 and 8. The points a, b, c, and d respectively represent maximum peaks of transition state (S)TS and (R)TS in IR vibrating intensity (shown in Tab. 1 and 2 in the supporting information).
As for transition state (S)TS to be illustrated, at the point of a, it indicates that flexible vibration of proton H(25) originated form chiral quinine oriented to N(57) atom of benzothiazol imines. At the point of b, it identifies that flexible vibration of enolic diethyl malonate’s proton H(90) oriented to tertiary amine N(1) of quinine. All of data demonstrate that both of hydroxyl and tertiary amine N (1) from chiral quinine are active sites relying on strong flexible vibration of hydrogen bonding interaction. At the point of c, it agrees to flexible vibration of C(56)—N(58) and C(56)—N(57); At d point, it promises flexible vibration of C(80)—O(79). In both of c and d points data, the hydroxyl O(12)—H(25) and tertiary amine N (1) attained from chiral quinine regarded as the active sites are affirmative further. Vibrating diagnosis for transition state (R)TS is similar to that of transition state (S)TS.
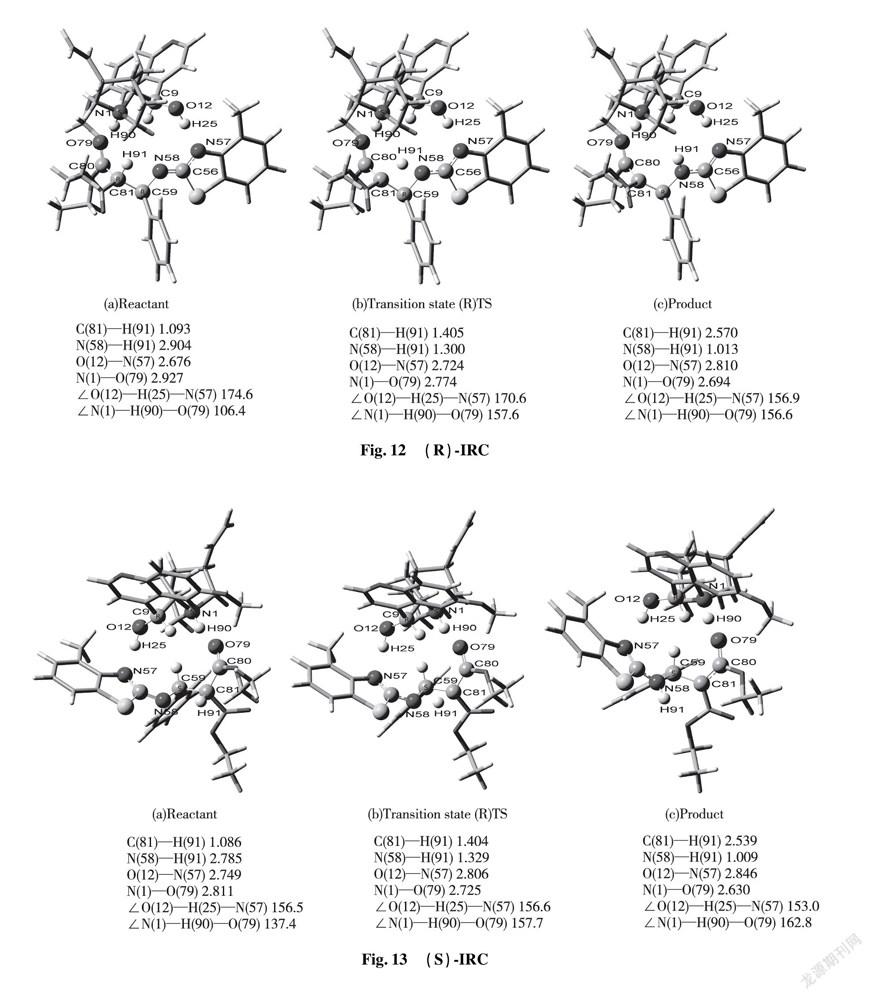
2.3.3 Main Parameters and Reaction Mechanism of Transition State Molecules Via the DFT calculation, we successfully confirmed the most stable transition sate of catalyst-substrates complexes through hydrogen bonding interaction. Theoretical calculation predicted that the stable transition state (R)TS and (S)TS are involved in formation of hydrogen bonds among Cat, EI and enolic Nu. In term of transition state (S)TS, the calculating bonding length of hydrogen bond N(1)—H(90)—O(79) is 2.725 Å; the angle of ∠N(1)—H(90)—O(79) is 157.7°, which represents strong hydrogen bond existed between tertiary amine of chiral quinine and enolic isomer of diethyl malonate. The calculating bonding length of hydrogen bond O(12)—H(25)—N(57) is 2.806 Å; the angle of ∠O(12)—H(25)—N(57) is 156.6°, that is, the information indicates formation of strong hydrogen bond. After combination among Cat, EI and Nu by hydrogen bonds, the length of diethyl malonate C(80)—C(81) is increased from 1.355 Å to 1.454 Å that achieves the range of C—C single bond’s formation. Simultaneously, C(81) of diethyl malonate attacked to C(59) of benzothiazol imine to form C—C single bond with the bonding length 1.552 Å. The double bond length between N(58)—C(59) is increased from 1.274 Å to 1.469 Å, which reached the range of C—N single bond formation. The proton H(91) placed in the middle of C(81) and N(58) produces vibration. The distance of C(81)—H(91) and N(58)—H(91) are 1.404 Å and 1.329 Å respectively; the angle of ∠N(58)—H(91)—C(81) is 111.0° in Fig.9.
The diagnosis of transition state (R)TS is similar to that of transition state (S)TS. The hydrogen bonding length N(1)—H(90)—O(79) is 2.774 Å, close to that of O(12)—H(90)—N(57) with 2.724 Å. Both of corresponding angles ∠N(1)—H(90)—O(79) and ∠O(12)—H(25)—N(57) are 157.6° and 170.6° respectively.
2.4 Result of IRC Analysis
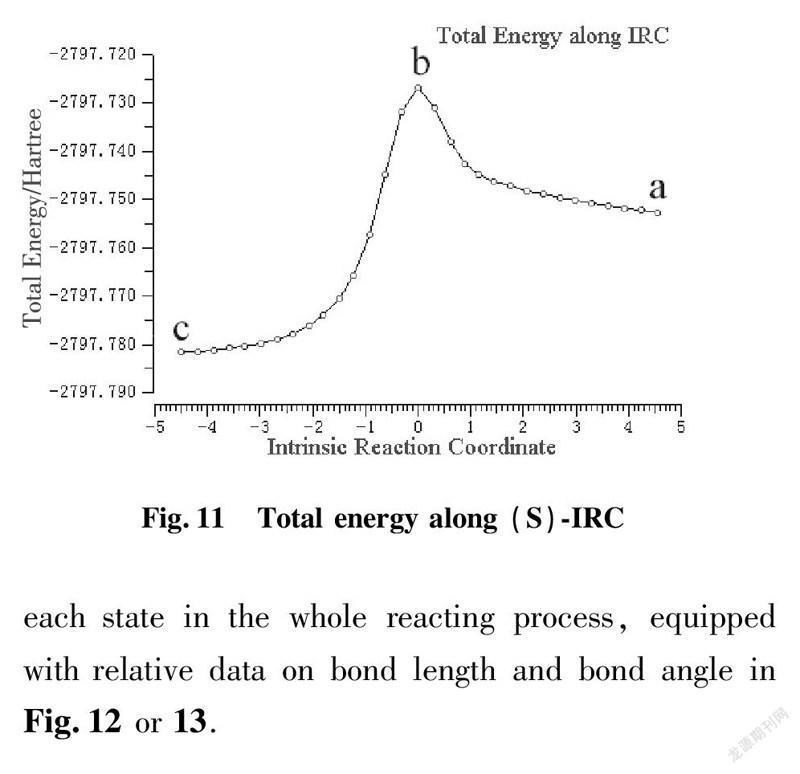
To illustrate reacting mechanism with transition state, we applied the method of IRC to explore further relationship among reactant, transition state and product at the same level of basis set as shown in Fig.10 and 11.
a is beginning state of reaction; b is transition state; c is terminal state of reaction
IRC approach is applied to kinetic diagnosis for each state in the whole reacting process, equipped with relative data on bond length and bond angle in Fig.12 or 13.
3 Conclusions
In this paper, we have investigated mechanism of asymmetric Mannich reaction of benzothiazol-β-amino esters catalyzed by chiral quinine organocatalyst (Cat). The main conclusions drawn from our investigation are summarized as follows:
1) Chiral quinine organocatalyst containing four chiral carbons performed specific geometry, that is the determination of geometric product.
2)Via DFT calculation for transition state, the active sites of chiral quinine organocatalyst are hydroxyl and tertiary amine, which had been confirmed.
3)To investigate further reacting mechanism of Mannich reaction in the presence of chiral quinine organocatalyst: Firstly, diethyl malonate is converted into its enolic isomer as nucleophile to be hydrogen bonding donor, and then it is combined with tertiary amine of quinine organocatalyst through hydrogen bond. At the same time, benzothiazol imine N (57) is connected to hydroxyl of chiral quinine organocatalyst by hydrogen bond to collide with diethyl malonate combined with tertiary amine of chiral quinine organocatalyst by hydrogen bond to afford relating optical product.
4)Since transition state (S)TS is lower than (R)TS in energy, the product is obtained mainly with S configuration.
5)Controlling reacting temperature is one of the important factors for increasing distereoselectivity. Beforehand assuring reacting rate, the lower temperature is going on, the more enantio-and distereoselectivity are upgraded.
Acknowledgments:We are grateful for financial support from the Science Foundation of Guizhou Province([2017]1028). We also acknowledge the key surper-computing chemistry lab of Guizhou Province.
References:
[1]ZHU J L, ZHANG Y, LIU C, et al. Insights into the dual activation mechanism involving bifunctional cinchona alkaloid thiourea organocatalysts: an NMR and DFT study[J]. The Journal of organic chemistry, 2012,77(21):9813-25.
[2] CHAUHAN P, CHIMNI S S. Asymmetric addition of indoles to isatins catalysed by bifunctional modified cinchona alkaloid catalysts[J]. Chemistry-A European Journal, 2010,16(26): 7709-13.
[3] ALESSIO R, ALESSANDRA L. Asymmetric organoca
talytic conjugate addition of diarylphosphane oxides to chalcones[J]. European Journal of Organic Chemistry, 2010, 2010(35):6736-6739.
[4] BUSYGIN I, NIEMINEN V, TASKINEN A, et al. A combined NMR, DFT, and X-ray investigation of some cinchona alkaloid O-ethers[J]. The Journal of organic chemistry, 2008,73(17):6559-69.
[5] WANG W, ZHANG G P, SONG B A, et al. Synthesis and anti-tobacco mosaic virus activity of O,O'-Dialkyl-a-(substituted benzothiazol-2-yl)amino-(substituted phenyl
methy1)phosphonate[J]. Chin J Org Chem, 2007, 27(2): 279-284.
[6] LONG N, CAI X J, SONG B A, et al. Synthesis and antiviral activities of cyanoacrylate derivatives containing an α-aminophosphonate moiety[J]. J Agric Food Chem, 2008, 56: 5242-5246.
[7] HU D Y, WAN Q Q, YANG S, et al. Synthesis and antiviral activities of amide derivatives containing the α-aminophosphonate moiety[J]. J Agric Food Chem, 2008, 56(3): 998-1001.
[8] LI L, SONG B A, BHADURY P S, et al. Enantioselective synthesis of β-amino esters bearing a benzothiazole moiety via a Mannich-type reaction catalyzed by a cinchona alkaloid derivative[J]. Eur J Org Chem, 2011: 4743-4746.
[9] BAI S, LIANG X P, SONG B A, et al.Asymmetric Mannich reactions catalyzed by cinchona alkaloid thiourea: enantioselective one-pot synthesis of novel β-amino ester derivatives[J]. Tetra: Asym, 2011, 22: 518-523.
[10]McLean A D, Chandler G S.Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z=11-18[J].J Chem Phys, 1980, 72: 5639-5648.
[11]RAGHAVACHARI K, BINKLEY J S, SEEGER R, et al.Self-consistent molecular orbital methods. 20. basis set for correlated wave-functions[J].J Chem Phys, 1980, 72: 650-54.
[12]TUMA C, BOESE A D, HANDY N C.Predicting the binding energies of H-bonded complexes:a comparative DFT study[J]. Phys Chem Chem Phys, 1999,1: 3939-3947.
[13]RABUCK A D, SCUSERIA G E. Performance of recently developed kinetic energy density functionals for the calculation of hydrogen binding strengths and hydrogen-bonded structures[J].Theor Chem Ace, 2000, 104: 439-444.
[14]SHERER E C, YORK D M, CRAMER C J. Fast approximate methods for calculating nucleic acid base pair interaction energies[J]. J Comput Chem, 2003, 24: 57-67.
[15]XU X, GODDARD W A. Bonding properties of the water dimer: a comparative study of density functional theories[J].J Phys Chem, 2004, 108: 2305-2313.
[16]ZHAO Y, TRUHLAR D G. Benchmark databases for nonbonded interactions and their use to test density functional theory[J].J Chem Theory Comput, 2005, 1: 415-432.
[17]SUN T, WANG Y B. Calculation of the binding energies of diferent types of hydrogen bonds using GGA density functional and its long-range,empirical dispersion correction methods[J]. Acta Phys-Chim Sin, 2011, 27(11): 2553-2558.
[18]FUKUI K. The path of chemical-reactions-the IRC approach[J]. Acc Chem Res, 1981, 14: 363-68.
[19]HRATCHIAN H P, SCHLEGEL H B. Accurate reaction paths using a hessian based predictor-corrector integrator[J]. J Chem Phys, 2004, 120: 9918-24.
[20]FRISCH M J, TRUCKS G W, SCHLEGEL H B, SCUSERIA G E, ROBB M A, CHEESEMAN J R, SCALMANI G, BARONE V, MENNUCCI B, PETERSSON G A, NAKATSUJI H, CARICATO M, LI X, HRATCHIAN H P, IZMAYLOV A F, BLOINO J, ZHENG G, SONNENBERG J L, HADA M, EHARA M, TOYOTA K, FUKUDA R, HASEGAWA J, ISHIDA M, NAKAJIMA T, HONDA Y, KITAO O, NAKAI H, VREVEN T, MONTGOMERY J A, PERALTA J E, OGLIARO F, BEARPARK M, HEYD J J, BROTHERS E, KUDIN K N, STAROVEROV V N, KOBAYASHI R, NORMAND J, RAGHAVACHARI K, RENDELL A, BURANT J C, IYENGAR S S, TOMASI J, COSSI M, REGA N, MILLAM J M, KLENE M, KNOX J E, CROSS J B, BAKKEN V, ADAMO C, JARAMILLO J, GOMPERTS R, STRATMANN R E, YAZYEV O, AUSTIN A J, CAMMI R, POMELLI C, OCHTERSKI J W, MARTIN R L, MOROKUMA K, ZAKRZEWSKI V G, VOTH G A, SALVADOR P, DANNENBERG J J, S DAPPRICH, A D DANIELS, O FARKAS, J B FORESMAN J V, CIOSLOWSKI O J, FOX D J. Gaussian 09(Revision A.02),Gaussian, Inc., Wallingford CT,2009.
(责任编辑:于慧梅)
作者简介:谢承卫(1964—),男,教授,博士,研究方向:手性合成反应机理,E-mail:cwxie@gzu.edu.cn.
通讯作者:谢承卫,E-mail:cwxie@gzu.edu.cn.
奎宁手性催化合成苯并噻唑氨基酸酯反应机理研究
摘 要:苯并噻唑-β-氨基酸酯不对称Mannich反應研究,对寻找具有良好生物活性的对映异构体具有重要意义。本文采用高精度量化计算和试验相结合的方法,研究了奎宁手性催化合成苯并噻唑-β-氨基酸酯的Mannich反应机理。利用密度泛函理论(DFT)M06-2X方法,溶剂采用CPCM模型,在6-311G(d,p)基组水平上,对反应的过渡态TS(S,R)进行了详细的研究。结果表明,奎宁手性催化作用位点有两个,分别形成O(12)—H(25)—N(57)和N(1)—H(90)—O(79) 氢键。通过过渡态红外振动频率计算与分析进一步验证了过渡态的准确性。计算结果与试验结果能很好吻合,反应体系的温度是提高立体选择性的关键因素之一,温度越低立体选择性越好。
关键词:奎宁;苯并噻唑-β-氨基酸酯;密度泛函;过渡态;反应机理
