钠通道NaV1.9与疼痛的研究进展
2021-01-09何询,周熙
何 询,周 熙
(1. 深圳未名新鹏生物医药有限公司,深圳 518057;2. 湖南师范大学生命科学学院动物多肽药物创制国家地方联合工程实验室,长沙 410081)
疼痛本是机体自我保护的一种机制,是一种重要的生理功能。然而在神经损伤、癌症、糖尿病、病毒感染以及某些炎症等病理条件下,疼痛会演变成慢性衰弱性疾病,即慢性疼痛,例如癌痛、糖尿病性神经痛、疱疹后神经痛等。保守估计,全球有将近五分之一的人常年遭受慢性疼痛的困扰[1]。目前临床上用于治疗疼痛的镇痛药物有非甾体类抗炎药、阿片类药物、局部止痛药、抗惊厥类药物、三环类抗抑郁药等[2]。尽管如此,上述镇痛药物仍存在专一性低、长期使用副作用大、具有成瘾性等问题,特别是近年来阿片类药物的滥用引发的不良社会问题日益尖锐。因此,新靶点新机制的镇痛药物的研发是生物和医药领域关注的热点方向。
电压门控钠通道在电信号的起始和传导中扮演了重要角色,其功能异常可能与多种慢性疼痛的发病机制相关,因此一直是镇痛药物研发的重要靶点[3]。在9个钠通道亚型中,电压门控钠通道NaV1.7、NaV1.8和NaV1.9特异性表达于外周神经系统(如背根神经节、三叉神经节等外周伤害感受神经元),调控外周疼痛信号通路[3]。临床前动物模型以及临床遗传学的研究表明,这3个钠通道在正常的生理以及病理性疼痛感受中扮演着重要角色。目前,有多个针对神经病理性疼痛的NaV1.7和NaV1.8的抑制剂正处于临床研究阶段[4-5]。据Cortellis(http://cortellis.thomsonreuterslifesciences.com)数据显示,现在共有40个NaV1.7抑制剂正处于临床研究阶段,其中Newron Pharmaceuticals公司研发的针对神经病理性疼痛的镇痛药物Ral finamide正处于临床三期研究阶段。然而,至今仍未有关于靶向NaV1.9的临床前或临床研究中的药物报道。本文将对NaV1.9与疼痛的关系以及靶向镇痛药物的开发进行综述和探讨。
1 NaV1.9的表达定位和生理学特性
在初级感觉神经元中分布有一系列特殊的离子通道和受体,这些离子通道和受体响应外界刺激(如化学、机械和温度等),引起局部的膜电位去极化,从而产生电信号将信息传入中枢。NaV1.9主要分布在无髓鞘的C-fiber末梢可被IB4标记的非肽能背根神经节(dorsal root ganglion, DRG)的神经元上(图 1),同时也在三叉神经节神经元和肠肌层神经元中高表达[6-7]。众多研究表明,NaV1.9参与了上述神经元电信号的产生与传导。
NaV1.9在亚细胞中的定位受蛋白质翻译后修饰和膜转运调控。辅助亚基β 1和β2能显著上调NaV1.9在异源细胞HEK293T质膜上的功能性表达[8]。体外试验发现,细胞黏附蛋白可直接作用于NaV1.9的C末端,并且上调NaV1.9在质膜上的表达;而当细胞黏附蛋白被敲除后,IB4标记的神经元中NaV1.9的电流密度显著降低,表明细胞黏附蛋白能直接与NaV1.9相互作用来促进NaV1.9的跨膜转运[9]。
相比于其他钠通道,NaV1.9具有独特的电生理特性。在静息膜电位时,NaV1.9能响应阈下刺激产生持续钠离子内流,使细胞膜去极化;而在动作电位时,其能作为“阈值通道”来影响神经元的兴奋性。Herzog等[10]通过计算机模拟发现,即使将DRG胞体中NaV1.9在细胞膜上的表达丰度降低到50%,其对细胞膜的去极化仍具有相当大的贡献(仍能大于正常水平时的75%)。因此,根据NaV1.9的组织分布特性和电生理特征,我们推测它在疼痛信号的产生和传导中应具有重要的作用。
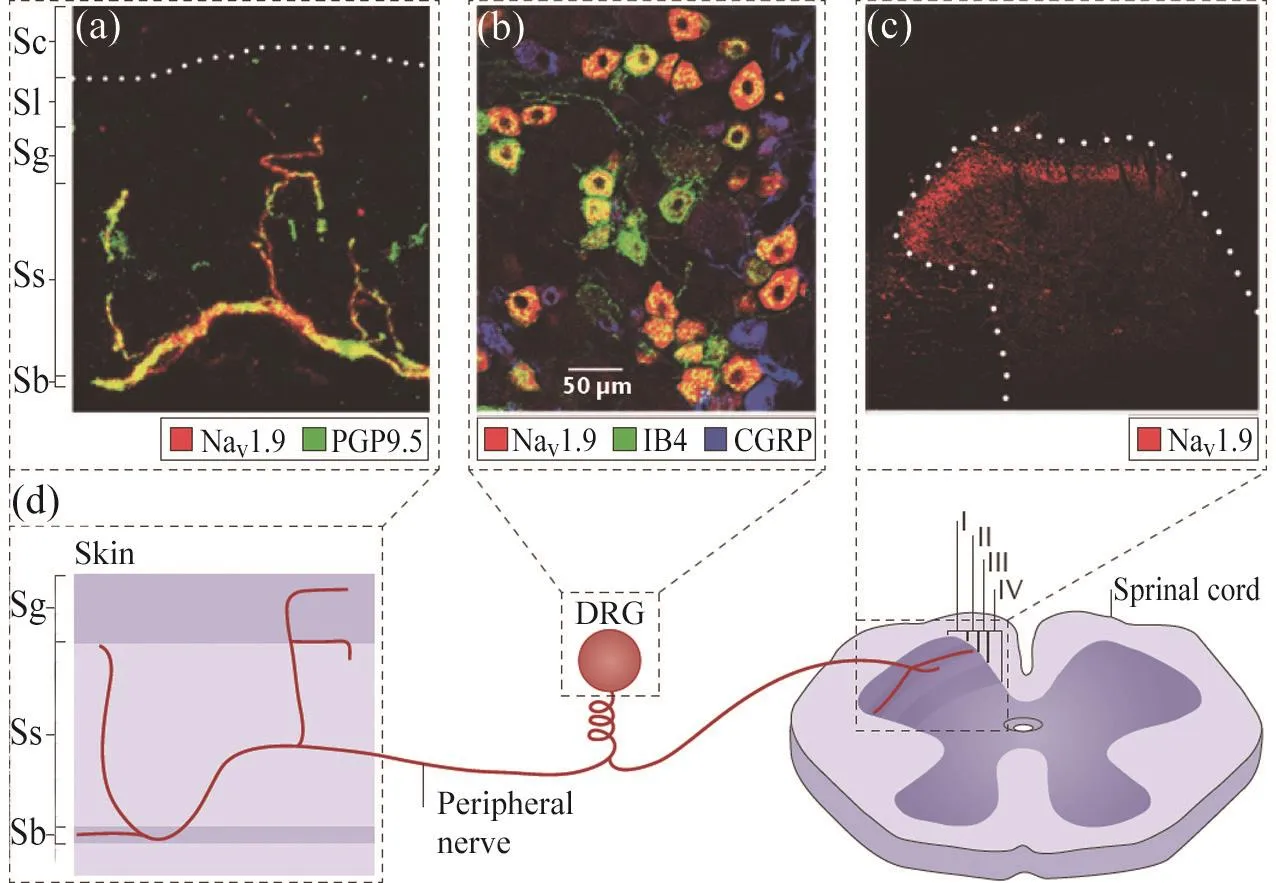
图1 NaV1.9在DRG神经元中的分布[6]Fig. 1 Distribution of NaV1.9 in DRG neurons[6](a)NaV1.9在表皮内神经纤维中的表达,其与蛋白基因产物PGP9.5存在共定位,图中黄色显示两者共定位;(b)NaV1.9在DRG神经元中与IB4存在非常高的共定位,但不与降钙素基因相关肽共定位;(c)脊髓后角浅层DRG神经元突触前神经末梢显示有NaV1.9的免疫染色;(d)NaV1.9在皮肤初级传入神经末梢、DRG胞体和中枢神经末梢分布的模式示意图。PGP9.5:蛋白基因产物9.5;CGRP:降钙素基因相关肽 ;DRG :背根神经节 ;Sc:角质层 ;Sl;透明层 ;Sb :基底层 ;Ss:棘皮层 ;Sg :颗粒层 ;Ⅰ:板层Ⅰ;Ⅱ:板层Ⅱ;Ⅲ:板层Ⅲ;Ⅳ:板层Ⅳ。(a) Expression of NaV1.9 in intraepidermal nerve fibres, colocalization of NaV1.9 and PGP9.5 is shown in yellow; (b) DRG neurons display substantial colocalization of NaV1.9 with the isolectin IB4 (green) but not with calcitonin gene-related peptide (CGRP, blue); (c) Presynaptic nerve terminals of DRG neurons in the super ficial layers of the dorsal horn of the spinal cord display NaV1.9 immunostaining; (d) A schematic representing the localization of NaV1.9 channels at primary afferent peripheral nerve endings in the skin, in DRG somata and in central nerve endings. PGP9.5: Protein gene product 9.5; CGRP: Calcitonin gene-related peptide; DRG: Dorsal root ganglion; Sc: Stratum corneum;Sl: Stratum lucidum; Sb: Stratum basale; Ss: Stratum spinosum;Sg: Stratum granulosum; Ⅰ: Lamina Ⅰ; Ⅱ: Lamina Ⅱ; Ⅲ: Lamina Ⅲ;Ⅳ: Lamina Ⅳ.
2 NaV1.9在疼痛中的作用
2.1 动物模型试验
野生型小鼠在炎症反应后会对机械和温度产生超敏,然而NaV1.9基因敲除小鼠在炎症反应后对机械和温度刺激的敏感度显著下降,这表明 NaV1.9 可能参与了炎症的超敏反应[11]。另一项动物模型试验表明NaV1.9参与炎性疼痛。当大鼠后肢产生炎症反应后,支配其后肢的DRG感觉神经元中NaV1.9的表达增加[12]。炎性疼痛时,NaV1.9功能上调的现象存在2种可能的机制:1)炎性因子增加,促使NaV1.9功能上调;2)炎症引起胆固醇含量降低,促使NaV1.9功能上调。当组织损伤时,损伤细胞释放大量的炎性因子,如 H+、缓激肽、组胺、三磷酸腺苷(adenosine triphosphate, ATP)、三磷酸鸟苷 (guanosine triphosphate,GTP)、五羟色胺、神经营养因子、前列腺素2等。这些炎性因子可直接或间接增强NaV1.9的功能,降低动作电位爆发的阈值,从而增强神经元的兴奋性,并且在给予相同的去极化刺激时,炎性因子处理的DRG神经元的动作电位爆发频率增加[13]。在产生炎性因子的同时,炎症反应也降低了局部胆固醇的含量。胆固醇含量的降低可以使伤害感受神经元敏感化,促进机械和热痛觉过敏。Amsalem等[14]通过NaV1.9敲除小鼠动物模型和细胞电生理试验证明了胆固醇含量降低能激活NaV1.9而增强神经元的兴奋性,从而对机械刺激和热痛觉产生超敏反应。
Luiz等[15]发现,NaV1.9在三叉神经痛相关的神经性口面部疼痛的发展中起着重要作用。敲除NaV1.9并不影响小鼠正常的面部疼痛行为(机械和热刺激),然而在面部眶下神经压迫(constriction of the infraorbital nerve, CION)的神经性疼痛模型(该模型诱导面部超敏的神经性疼痛,并且可以导致长达3周对机械刺激超敏感)中,相对于野生型小鼠,NaV1.9敲除小鼠的面部区域对机械和热刺激的敏感性显著下降,并且也去除了模型小鼠的持续性机械超敏感。
长期或过量使用一些非处方或镇痛药(如扑热息痛、类阿片和联合镇痛药)将可能导致药物过度使用性头痛(medication-overuse headaches, MOH)。Bonnet等[16]发现这可能与NaV1.9存在一定的关系。他们发现治疗偏头痛的曲坦类药物导致的MOH症状和一氧化氮(NO)异常激活NaV1.9有关。当敲除NaV1.9的基因后,MOH症状小鼠未表现出NO介导的症状,这些症状包括头皮异常性疼痛、光和声音恐惧症。同时,NaV1.9的非正常激活能触发降钙素基因相关肽(calcitonin gene-related peptide, CGRP)的释放,引起动脉扩张和肥大细胞脱颗粒。反过来,肥大细胞释放的炎性因子能促进NaV1.9表达上调,从而加重炎症和疼痛信号。
Lolignier等[17]发现,NaV1.9在冷刺激引发的疼痛感受中扮演了重要的角色。在有害冷环境中,冷敏感的感觉神经元中NaV1.9电流上调,阈下刺激放大,神经元放电增加,从而导致冷痛。在NaV1.9敲除的小鼠中,冷刺激诱发的冷痛和奥沙利铂化疗后的疼痛性冷超敏反应均有所降低。
另外,也有研究指出,在大鼠骨癌痛模型中,DRG神经元中NaV1.9的mRNA水平和蛋白质水平在肿瘤的不同发展阶段较对照组均有显著的上升,表明NaV1.9 可能参与了骨癌疼痛的发展与维持[18]。
综上所述,动物模型研究表明,NaV1.9在炎性疼痛、神经性疼痛和冷痛中均扮演了重要角色。
2.2 人类临床相关疾病的研究
近些年来,高通量测序技术的发展加速了人类对罕见遗传疾病的研究。在人类的疼痛研究中,一些罕见的遗传性疼痛和外周神经痛疾病与编码NaV1.9的基因SCN11A的突变相关[6]。目前共鉴定有18个不同位点的突变,可划分为3类不同的疼痛疾病:1)先天性无痛症;2)家族性发作性疼痛;3)小纤维神经痛。2013年,德国科学家Leipold等[19]首次在Nature Genetics上报道,NaV1.9功能获得性突变L811P能导致患者先天性无痛症,该患者不能感知有害的疼痛刺激,并伴随有自残行为和无痛性骨折。同年,我国学者刘静宇教授、姚镜教授及张学教授等[20]合作的研究发现,2个发作性疼痛家系分别与NaV1.9的R225C和A808G突变相关;功能学研究表明,2个位点的突变均增加了DRG神经元的兴奋性。2014年,Huang等[21]对345个外周神经病理性疼痛散发患者的疼痛相关基因进行了筛查,发现11个病例的SCN11A基因发生突变,共涉及8个不同位点的突变。作者对其中2个突变I381T和L1158P进行了功能性分析,发现突变通道可导致激活曲线向超极化方向漂移,增大了窗口电流,并使去激活变慢。电流钳试验发现,这2个突变增加了DRG神经元动作电位的自发性爆发,也增加了诱发的动作电位频率。这3个研究工作在NaV1.9遗传学研究中具有代表性,拉开了深入研究NaV1.9在疼痛中角色的序幕。因此,基于上述人类遗传学的研究充分证明了NaV1.9在痛觉产生及痛觉异常的疾病中发挥了重要作用。
2019年,Salvatierra等[22]发现NaV1.9可能在瘙痒中也扮演了重要角色。他们发现该通道L811P突变的病人虽表现出对疼痛不敏感,但却伴随有严重的瘙痒行为。作者随后构建同源突变敲入小鼠(NaV1.9L799P/WT)和NaV1.9敲除小鼠对其进行了研究,发现致痒原引起了野生型小鼠强烈的急性瘙痒反应,NaV1.9敲除小鼠致痒原引起的急性瘙痒行为却显著降低。然而,NaV1.9L799P/WT小鼠在安静状态下表现出比野生型小鼠更多的自发性瘙痒。通过细胞试验表明,致痒原(如组胺)引起瘙痒的原因可能是其增强了NaV1.9的活性从而改变了动作电位的特性。在另一项研究中,Zhou等[23]发现组胺可直接增强NaV1.9电流,说明组胺不仅可作为炎性因子激活NaV1.9致痛,也可作为致痒原激活NaV1.9导致瘙痒。这些研究表明,NaV1.9可能是治疗瘙痒的一个药理学靶点。
3 NaV1.9的药理学研究
动物模型和临床遗传学研究表明,NaV1.9在疼痛信号通路中扮演了重要角色,是一个潜在的镇痛药物开发靶点。但相对于其他钠通道亚型,NaV1.9的研究要滞后得多,其功能和药理学相关的直接证据相对较少,极大地限制了其药理学研究和新药研发的进展。这主要是因为长时间以来NaV1.9无法实现异源细胞的功能性表达,缺少药理学调制剂,难以通过电生理试验对其功能和药理学进行系统研究[6]。近几年来,共有4个不同的NaV1.9异源表达体系被建立,其能在如HEK293、ND7/23和CHO-K1等异源细胞上表达出幅度大且稳定的电流。利用建立的异源表达系统,结合动物模型等研究,Zhou等[24]从白额巨蟹蛛毒液中发现蜘蛛的多肽毒素HpTx1能激活NaV1.9,增强神经元的兴奋性,从而诱发小鼠的伤害感受疼痛行为,降低机械痛和热痛阈值。值得注意的是,HpTx1通过激活NaV1.9而逆转NaV1.7敲除小鼠的无痛反应这一发现可能为治疗NaV1.7相关的先天性无痛症提供新策略和新靶点,为相关药物研发提供新思路。同时该研究也为NaV1.9 在疼痛感知中的重要角色提供了药理学证据支持。此外,Zhou等[24]进一步分析了蜘蛛多肽毒素HpTx1作用于NaV1.9通道上的关键位点,发现它是通过与NaV1.9的第4个电压敏感结构域相结合来阻碍通道快失活的,说明NaV1.9类似于另外8种亚型,其第4个结构域跟通道快失活相关。因此,该研究也表明NaV1.9具有与其他钠通道亚型相类似的多肽毒素结合区域和功能特征。
Liang等[25]研究发现,阿密曲替林(amitriptyline,一种三环类抗抑郁药,也对偏头痛有很好的治疗效果)对三叉神经节中NaV1.9电流也有阻断作用,能使稳态失活曲线向超极化方向显著地漂移且不影响稳态激活,这样将导致通道更容易失活而抑制NaV1.9电流。
Lin等[8]发现多个化合物(包括阿密曲替林、利多卡因、苯唑卡因、布比卡因、氟卡尼、拉莫三嗪、美西律、TC-N 1752、丁卡因和尼莫地平)对NaV1.9均有抑制作用。当NaV1.9上相对应的经典局部麻醉剂作用位点F1592和Y1599双突变成丙氨酸时,阿密曲替林、利多卡因、苯唑卡因、美西律、TC-N 1752和丁卡因对突变后通道的亲和力均有不同程度的下降,其中利多卡因和TC-N 1752的亲和力下降约100倍。它表明这些化合物相互作用于通道第4个结构域的第6个跨膜片段内环上经典的局部麻醉剂结合位点。由此说明,NaV1.9跟其他钠通道一样,可被广谱活性的局部麻醉类药物抑制。
4 总结与展望
综上所述,NaV1.9在炎性疼痛、神经病理性疼痛和冷痛中发挥了重要作用,也可能在癌痛和内脏痛中有一定作用。NaV1.9在组织表达中的特异性和一级序列的独特性赋予了其成为开发特异性作用药物靶点的潜力,可降低药物脱靶而导致的副作用。再者,所有发现的NaV1.9遗传突变病例除了有疼痛紊乱的症状外,并没有其他如智力和认知等方面的问题,故可推测靶向NaV1.9通道不会引起不良的认知缺陷。目前研究也表明,多肽毒素或炎性因子等能增强NaV1.9功能而诱发疼痛或疼痛加剧,这从药理学方面揭示了NaV1.9与疼痛的重要关系。因此,NaV1.9可能是一个较为理想的潜在镇痛药物靶点,其特异性抑制剂可能不会对心脏、骨骼肌和认知等功能产生影响。然而有报道发现,NaV1.9在大鼠下丘脑视上核大神经分泌细胞中有表达,其可能对该细胞的放电有贡献[26],说明在药物研发中,NaV1.9抑制剂(特别是可穿透血脑屏障的抑制剂)对下丘脑功能的影响是需要谨慎对待和评估的。此外,有报道也表明,NaV1.9在肠神经元中有高表达,参与肠神经元的兴奋性,其功能上调可增强肠传入神经的放电,导致肠神经元的兴奋性增加,肠组织机械超敏[27]。这在一定程度上说明NaV1.9可能与肠道疼痛相关,特别是临床上部分NaV1.9突变的患者存在不同程度的肠道功能紊乱(如腹泻、便秘、疼痛等)的现象也为该观点提供了一定的支持。但NaV1.9与内脏痛的关系仍不明确,其参与内脏痛的作用分子机制仍不清楚,同时抑制NaV1.9对肠道正常生理功能的影响也不清楚,需要进一步的系统研究。总的来讲,NaV1.9调控外周疼痛信号,是一个潜在的镇痛药物研发靶点,但仍充满未知,机遇与挑战并存。
基于目前的研究现状,靶向NaV1.9的药物研发还任重道远。目前还没有高亲和力和高专一性的NaV1.9调制剂,需要加大筛选力度。蜘蛛、蜈蚣、蝎子等有毒动物富含多肽毒素,它们是有毒动物在亿万年进化过程中的产物,具有惊人的分子多样性、结构多样性和功能多样性,是新药研发的重要资源宝库,也是当前新药筛选的一个重要方向。此外,某些对钠通道具有状态依赖性的小分子抑制剂具有较高的专一性和亲和力(如现在多个进入临床的NaV1.7小分子抑制剂)[4],因此,状态依赖性的小分子筛选也是一个重要的方向。当前,随着结构生物学的快速发展,特别是冷冻电镜在结构生物学中的应用,之前难以解析的膜通道蛋白现在可以呈现近原子或原子水平的清晰三维结构,为基于结构的药物设计提供了良好的模板和分子基础。目前已有4个哺乳动物钠通道亚型(NaV1.2、NaV1.4、NaV1.5和NaV1.7)的结构被解析。基于此,通过分子动力学模拟结合人工智能、计算机辅助药物设计等技术筛选和设计靶向NaV1.9的药物将成为可能,这也是靶向NaV1.9药物研发的一个重要方向。所以,在现在已具备NaV1.9异源表达体系的情况下,多方面资源与技术的结合将会加速作用于NaV1.9调制剂的筛选,促进靶向NaV1.9的药物研发。
