基于随机森林算法预测减压馏分油中噻吩硫化物的组成分布
2021-01-04任小甜褚小立田松柏
任小甜, 褚小立, 田松柏
(中国石化 石油化工科学研究院,北京 100083)
减压馏分油(VGO)是目前重油加工最主要的原料之一。一般而言,VGO中含有各种类型的硫化物,其中噻吩类硫化物主要有苯并噻吩、二苯并噻吩、萘苯并噻吩等。目前,VGO中硫化物的组成分布主要采用GC-MS方法分析,操作复杂、时间长,造成生产工艺参数调整的延迟。实现VGO中不同类型噻吩硫化物组成分布的及时预测分析,有助于VGO加工工艺中各项参数的实时优化和调整。相对而言,VGO的基本物性数据易于进行实时分析,且在生产过程中也积累了大量的物性分析数据,因此,构建通过VGO基本物性数据预测其噻吩类硫化物组成的模型,从而实现VGO中噻吩硫化物的组成分布的实时预测具有重要的意义。
近年来,已有一些基于石油馏分基本物性数据计算和预测其烃类组成的方法,基本思路是由馏分油基本物性数据计算和转换得到可以区分不同烃族的特征参数,然后以特征参数构建线性回归模型并进行预测。Riazi等[1-3]利用石油馏分的相对分子质量、折光指数、黏度以及密度等物性构建了不同的烃特征参数,并用其平均值代表对应的烃族,建立各物性的线性方程组,通过求解方程组来计算其中烷烃、环烷烃、芳烃以及硫化物的含量。邢波等[4]根据VGO的常规物性构建了5个新的特征参数,并选取80组VGO样本进行关联计算,通过求解带约束的最小二乘规划问题获得各项特征参数的系数,确定了VGO中链烷烃,单环和多环环烷烃,单环、双环和多环芳香烃含量的计算关联式。
以上方法需要先确定能区分不同烃族的特征参数,其本质是消除原始物性变量间的多元共线性,因此只能用于族组成种类较少的情况,对于详细的烃组成则找不到相应的特征参数。因此,除构造有物理意义的特征参数以外,还可将基本物性两两组合来构造新的特征参数。刘四斌等[5]收集了27个VGO样品的基本物性和族组成数据,并由其中5个基本物性(密度、折光率、相对分子质量、硫含量和氮含量)两两组合构建了15个回归因子,通过逐步回归法从中选择显著变量,分别建立预测13种烃类化合物组成的线性回归模型,其中总噻吩模型的预测精度最高,其显著变量为硫含量和折光率的乘积,表明VGO的总噻吩含量与其硫含量和折光率有较强的相关性。
除了线性回归模型外,利用非线性回归模型也可以直接拟合VGO族组成与其基本物性之间的关系。孙仁金等[6]利用动量-自适应学习率的BP人工神经网络(ANN)构建了由VGO的平均沸点、密度、相对分子质量和折光率预测其饱和烃含量的非线性预测模型;并进行了深入研究,添加VGO的运动黏度作为输入特征,建立了预测其饱和烃含量的GA-ANN模型;此外,利用遗传算法(GA)确定隐含层神经元数量和学习率,可以提高模型预测的准确度[7-8]。Stratiev等[9-10]则建立了由VGO的平均沸点、密度和结构特征参数(由经验公式计算得到)分别预测其饱和烃加单环芳烃的总含量、多环芳烃含量、总芳烃含量的多项式回归模型。非线性回归模型不需要构造新的物性特征参数,适用范围更广,预测准确度更高。
目前,研究主要集中在VGO烃族组成的预测,还未见预测VGO中不同类型噻吩类硫化物组成的报道。因此,笔者从VGO的基本物性出发,采用随机森林回归算法构建模型,分别预测其中苯并噻吩、二苯并噻吩、萘苯并噻吩以及总噻吩的质量分数。
1 模型的构建
1.1 算法原理

用RFR建立的模型训练时间短,不需要进行特征数据的预处理,而且还可以计算出每个特征的重要程度用于特征选择;同时,模型对离群的异常样本不敏感,稳健性好,不易过拟合,有较强的泛化能力,能保证较高的准确度。
1.2 构建数据集
收集160个直馏VGO样本,切割自不同基属的国内外原油(包括塔河原油、胜利原油、科威特原油和俄罗斯原油等)。测定其各项基本物性数据,包括20 ℃密度(ρ20),碳、氢、硫元素的质量分数(w(C)、w(H)、w(S)),70 ℃折光率n70,平均相对分子质量M,体积平均沸点TV,40 ℃、80 ℃和100 ℃下的运动黏度(v40、v80、v100),黏度指数VI和相关指数BMCI。依照标准方法ASTM D3239测定VGO中不同类型噻吩类硫化物(包括苯并噻吩、二苯并噻吩和萘苯并噻吩硫物)的含量,并计算得到总噻吩的含量。汇总这160个样本的基本物性和噻吩硫化物的组成数据,构造数据集。对数据集进行随机划分,其中75%的样本(120个)作为训练集,用于随机森林回归模型的训练和调参;其余25%的样本(40个)作为测试集,用来评估模型的准确性和泛化能力。
1.3 模型构建
利用Python进行模型的编写,其中调用开源的机器学习库Scikit-Learn[12]实现随机森林回归算法。RFR算法属于非参数模型,其对样本数据的总体分布没有先验的假设,直接通过算法拟合待测性质与变量之间的非线性关系。构建模型时,先对训练集样本进行有放回的抽样得到若干个样本子集,然后在每个样本子集上构造回归树;对树中的每个非叶节点进行分裂时,随机选择若干个特征数进行计算。当每个子集都构造起相应的回归树时,即可建立起不同特征下相应的划分规则和取值,整个随机森林回归模型也就构建完成。
将VGO的12项基本物性作为模型输入特征X, 3种噻吩硫化物和总噻吩的质量分数依次作为输出变量y,构建4种随机森林回归预测模型,分别为:苯并噻吩质量分数的预测模型M1、二苯并噻吩质量分数的预测模型M2、萘苯并噻吩质量分数的预测模型M3和总噻吩质量分数的预测模型M4。然后,确定4种模型的超参数,进行模型的训练和调参,并评估其预测的准确性和泛化能力。
1.4 超参数寻优
在对随机森林回归模型进行训练之前先要确定模型的超参数。有2种超参数需要寻优,第一种是模型的框架参数,即抽样得到的样本子集个数,也就是构造回归树的数量NT。一般来说,NT太小则模型训练不足而容易欠拟合,预测准确度较差;NT太大则模型预测的准确度又会降低,计算量也会增加。因此,需要选择一个适中的数值。第二种需要寻优的参数是回归树参数,包括树的最大深度、叶节点中的最少样本数、叶子节点的最高数量、每个非叶节点划分使用的最大特征数MF等。由于本研究的样本数量和特征数量都较小,只需要对回归树的每个非叶节点划分使用的最大特征数MF进行调参,其他回归树的参数均设为默认值即可。本研究以训练集样本的袋外估计为基础进行调参,省去了繁琐的交叉验证计算,先对回归树的数量NT进行寻优,然后再调节最大特征数MF。选择不同的超参数,设定其取值范围,然后计算对应模型中每个袋外样本的预测值,以所有袋外样本的预测标准偏差(RMSE_OOB)为评价指标,当其取值最小时对应的超参数即为最优。
2 结果与讨论
2.1 模型的超参数
对M1、M2、M3和M4依次进行超参数的寻优计算,分别得到最优的回归数的数量NT和最大特征数MF。苯并噻吩质量分数预测模型M1的调参过程如图1所示。由图1可知:M1袋外样本的预测标准偏差(RMSE_OOB)随着回归树数量(NT)的增加先减小后增大,当NT为160时其取得最小值(图1(a));且RMSE_OOB随着最大特征数(MF)的增大而减小,当MF为12时其取得最小值(图1(b))。由此,可以确定该模型的最优超参数NT和MF分别为160和12。类似地,依次对其他3种模型进行调参,汇总4种模型的最优超参数得到:二苯并噻吩质量分数预测模型M2的最优超参数为:NT=60,MF=6;萘苯并噻吩质量分数预测模型M3的最优超参数为:NT=130,MF=10;总噻吩质量分数预测模型M4的最优超参数为:NT=285,MF=12。
2.2 模型的训练
确定了各模型的最优超参数之后,分别在对应的120个训练集样本上进行模型的训练计算,利用训练集样本的校正标准偏差(RMSEC)和拟合决定系数R2来评价模型的准确性和拟合效果,由袋外得分可初步评价模型的泛化能力。各模型中训练集样本的噻吩硫化物的组成分布范围和模型评价指标见表1。由表1可知,4种模型中噻吩硫化物的组成分布范围较宽,模型具有较好的代表性;利用随机森林回归算法可以实现对直馏VGO中3种噻吩硫化合物和总噻吩质量分数的准确预测,对于噻吩硫化物质量分数为0的样本,模型也能给出较准确的预测。这4种模型中,总噻吩质量分数预测模型M4的拟合效果最好,其拟合决定系数R2可达到0.993,萘苯并噻吩模型M3的拟合效果较差,拟合决定系数R2为0.976;RMSEC值的大小和噻吩硫化物质量分数的大小呈正相关关系,因而总噻吩质量分数预测模型的RMSEC值最大,但各模型的RMSEC值均小于标准分析方法(ASTM D3239)中的重复性要求,说明模型的拟合效果都较好。

图1 苯并噻吩质量分数预测模型的超参数优化Fig.1 Optimization of hyper-parameters of the prediction model of benzothiophenes mass fractions(a) Number of trees in the forest; (b) Max features used for the split
从袋外得分OOB_score来看:总噻吩质量分数预测模型M4的得分最高,达到0.948,说明其具有较好的泛化能力,能对训练集之外的样本作出较准确的预测;而萘苯并噻吩质量分数预测模型M3的得分最低,只有0.826,同样其对应的训练集样本的拟合效果也最差(R2=0.976),说明该模型的准确性和泛化能力都较低。
VGO中总噻吩质量分数的线性回归预测模型[5]的预测校正标准偏差RMSEC为0.580%,拟合决定系数R2为0.984。与之相比,本研究构建的总噻吩质量分数RFR预测模型的准确性更高,RMSEC为0.264%。而且,其他3种模型预测3种不同类型噻吩硫化物的质量分数的结果也较准确。
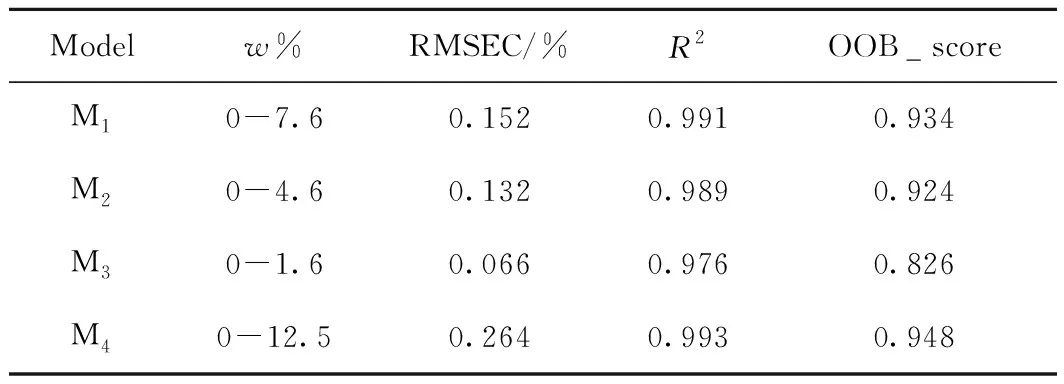
表1 各模型的噻吩硫化物组成分布范围和评价指标Table 1 The distribution range and evaluation index ofthiophene sulfides composition of each model
2.3 特征重要度分析
随机森林回归算法可以计算出模型中每个特征的重要程度。具体来说,在构造1颗回归树时,非叶节点按照分裂后的各个样本子集方差最小的准则进行分裂。这样可以计算出每个特征在某一节点处分裂前后的方差减少量,进而得到每个特征在所有回归树的相应节点上的平均方差减少量。将所有特征的方差减少量进行归一化计算,即得到每个特征的重要度。重要度表示每个特征在模型中的贡献值大小,可用于模型的特征筛选,即可从所有的12项物性特征中选择出若干个重要度较大的特征作为相应模型的主要特征,以这些特征为新的输入可以得到更简单的预测模型。
图2为各模型对应的特征重要度分析结果。由图2可知,这4种模型中硫质量分数特征的重要度最高,碳质量分数特征次之,而其他物性特征的重要度都很低,说明这些物性指标和噻吩硫化物组成的相关性很小。所以,各模型的重要特征均为硫质量分数(w(S))和碳质量分数(w(C))。在实际应用中,为了构建更加简单和轻量的VGO中噻吩硫化物质量分数的预测模型,可以只选择硫质量分数和碳质量分数作为输入特征来构建模型,也可以达到一定的预测准确度。
此外,由于减压馏分油中的硫基本上都分布在噻吩硫化物中,所以各模型中硫质量分数特征的重要度都最高;同时,VGO的各种噻吩硫化物中,苯并噻吩的含量最高,其占总噻吩含量的比例也最高;因此,对于苯并噻吩和总噻吩的2种模型(M1和M4),w(S)的特征重要度要远高于w(C);而二苯并噻吩及萘苯并噻吩的预测模型(M2和M3)中w(C)的特征重要度则相对较大,因为与苯并噻吩相比,这2种噻吩硫化物分子中分别增加6和12个C(分别为苯并噻吩分子中增加了1个和2个苯环);其结构中C的比例明显增大,使其硫化物质量分数与VGO中碳质量分数的相关性更大。

图2 各模型的特征重要度分布Fig.2 The feature importance distribution of each model(a) M1; (b) M2; (c) M3; (d) M4
2.4 测试集样本的预测分析
将测试集的40个样本数据分别代入这4种模型进行预测,利用测试样本的预测标准偏差RMSEP和拟合决定系数R2来评价各模型预测效果和泛化能力,并主要通过比较R2的大小来对比4种模型的预测准确性。测试集样本的3种噻吩化合物各自的质量分数及总噻吩质量分数的实测值和预测值对比如图3所示。从图3可知:苯并噻吩质量分数、二苯并噻吩质量分数和总噻吩质量分数的预测值和实测值基本吻合,表明模型M1、M2和M4的预测效果很好;萘苯并噻吩质量分数的预测值和实测值吻合度不好,有5个样本的预测偏差较大,说明模型M3的预测效果较差。同时,萘苯并噻吩模型的拟合决定系数最小,为R2=0.925,说明其拟合效果较差,泛化能力较弱;而苯并噻吩模型、二苯并噻吩模型和总噻吩模型的R2分别为0.963、0.988和0.981,说明苯并噻吩质量分数、二苯并噻吩质量分数和总噻吩质量分数预测模型的预测效果较好、泛化能力强。
分析萘苯并噻吩质量分数预测模型M3泛化能力较差的原因:由于随机森林回归算法不易产生过拟合现象,同时模型M3的训练集样本的拟合决定系数R2均小于其他3种模型,说明模型M3的自变量并不能很好地解释因变量的变化,即目前所选的12项物性特征不能充分反映VGO中萘苯并噻吩的组成信息,导致算法从现有数据集中学习得到的萘苯并噻吩质量分数和物性特征的映射关系不够准确,因此,对于测试集样本,其预测准确度较低,模型的泛化能力也较差。
选择模型的最重要特征硫质量分数(w(S))进行
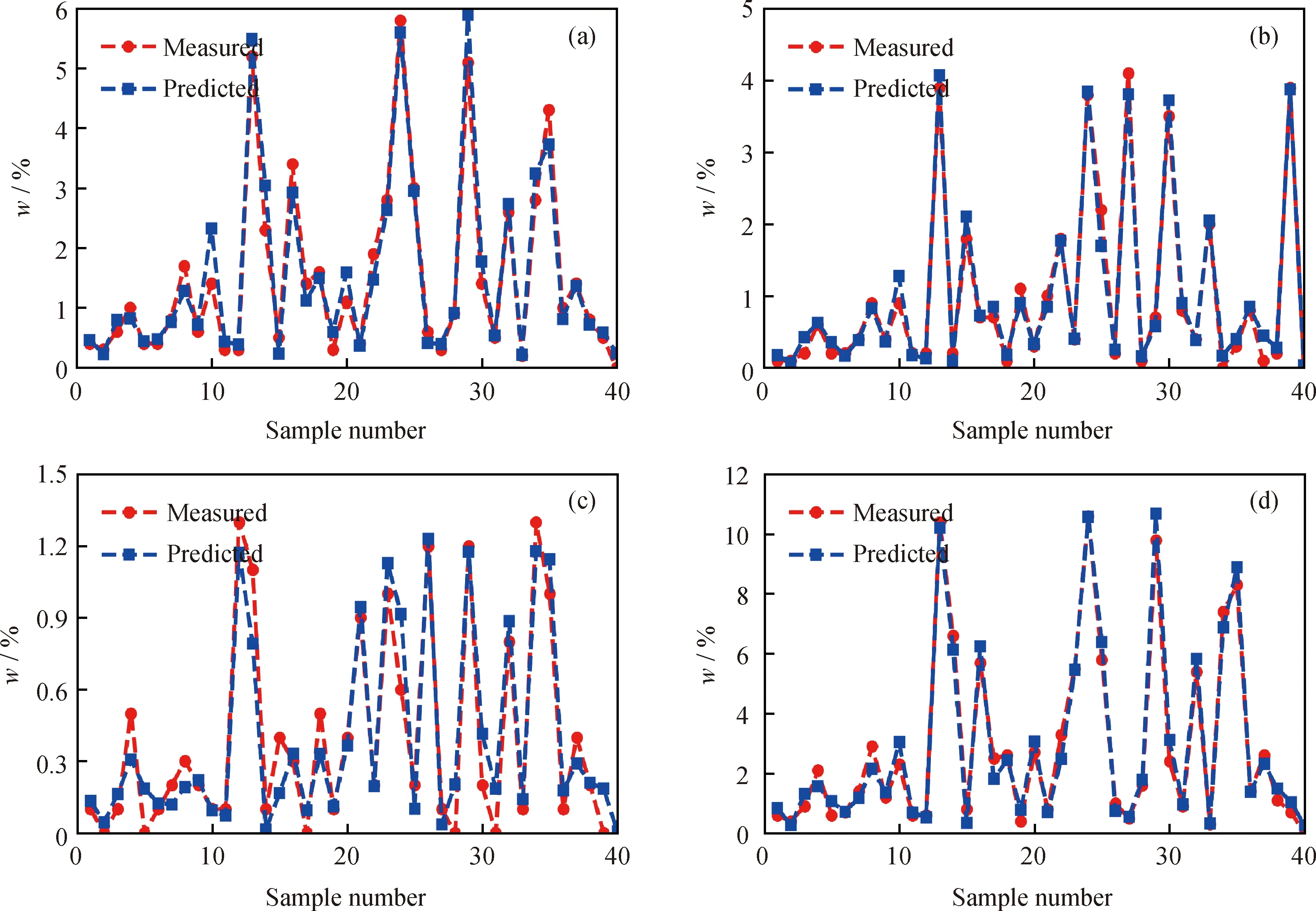
图3 测试集样本中噻吩硫化物组成的实测值和预测值对比Fig.3 Comparison of the measured and predicted values of thiophene sulfides’composition of the test set samples(a) Benzothiophenes; (b) Dibenzothiophenes; (c) Naphthathiophenes; (d) Total thiophenes
相关性可视分析,训练集样本中萘苯并噻吩质量分数与硫质量分数的关系如图4所示。由图4可知,VGO的萘苯并噻吩质量分数和(w(S))之间没有明显的相关关系,且存在硫质量分数相差较大的样本中萘苯并噻吩质量分数相同的情况。进一步计算VGO中萘苯并噻吩质量分数与12项物性特征(ρ20、w(C)、w(H)、w(S)、n70、M、TV、v40、v80、v100、VI和BMCI)的Spearman相关系数,结果分别为:0.58、-0.30、-0.69、0.80、0.62、-0.13、0.11、0.36、0.41、0.38、-0.32 和0.59。其中,w(S)的相关系数最高,为0.80;其他特征相关系数的绝对值均低于w(S)的。这说明VGO中萘苯并噻吩质量分数与上述12项物性特征的相关性都较差。因此,萘苯并噻吩质量分数预测模型的优化,需要筛选添加与其相关性好的VGO物性特征来优化预测模型,提高预测模型的准确度和泛化能力。
12个特征预测模型的预测评价结果列于表2。由表2可以看出,模型M1、M2、M3和M4的预测标准偏差(RMSEP)分别为:0.268%、0.131%、0.111%和0.385%。各模型的预测RMSEP值与其训练集样本中噻吩硫化物质量分数为正相关关系,即预测效果最差的萘苯并噻吩模型M3的RMSEP值最小,而预测最准确的总噻吩模型M4的RMSEP值则最大。3种噻吩硫化物预测模型的RMSEP值均小于标准分析方法(ASTM D3239)中数据的重复性要求:苯并噻吩0.8%、二苯并噻吩0.3%、萘苯并噻吩0.3%。这表明4种模型均能满足生产过程中快速分析和过程分析的要求。

图4 训练集样本的苯并噻吩质量分数和硫质量分数的散点图Fig.4 Scatter plot between the mass fraction ofnapathabenzthiophenes and sulfur of the train set samples
2.5 简化特征模型的构建与预测结果分析
为了提高模型的适用性,以硫质量分数(w(S))和碳质量分数(w(C))为输入特征重新构建模型,并对比不同特征数量模型的预测效果,结果如表2所示。由表2可以看出,对于3种噻吩硫化物和总噻吩的质量分数,只含2个特征预测模型的各项预测指标(袋外得分OOB_score、预测决定系数R2和预测标准偏差RMSEP)均略差于包含12个特征的预测模型,但也可以达到较高的预测精度,并且其RMSEP值同样均小于标准分析方法(ASTM D3239)中的数据重复性要求。

表2 不同数量特征的模型的评价指标Table 2 The evaluation index of the models with different number of features
上述结果表明,本研究构建的12特征预测模型和2特征预测模型都具有较强的泛化能力,能够准确预测训练集之外的样本。其中,2特征预测模型更加简便,仅由VGO的硫质量分数和碳质量分数就可以较准确预测3种噻吩硫化物和总噻吩的质量分数,实用性更强。
3 结 论
利用随机森林回归方法分别构建VGO中3种噻吩硫化物和总噻吩的质量分数预测模型,由直馏VGO的12项基本物性数据可以快速计算得到VGO中苯并噻吩、二苯并噻吩、萘苯并噻吩和总噻吩的质量分数,计算结果表明4种模型预测的准确性和预测结果的重复性较好,达到标准方法ASTM D3239的数据重复性要求,具有较强的泛化能力。
根据特征重要性的计算结果,选择VGO中硫质量分数和碳质量分数为输入特征构建了简化的预测模型,其预测结果也较准确,满足标准方法ASTM D3239的数据重复性要求,且更加简便、实用性更强。
