β2-AR别构拮抗剂Cmpd 15关键中间体(S)-9-芴基甲氧羰基-4-甲酰胺基苯丙氨酸的合成
2021-01-04钱明成
赵 帅, 芮 雪, 钱明成, 陈 新
(常州大学 药学院,江苏 常州 213164)
G蛋白偶联受体(G protein-coupled receptors, GPCRs)是体内最庞大的蛋白质超家族,目前已经有2000多种不同的GPCRs被报道[1-2]。G蛋白偶联受体通过与细胞周围环境中的配体结合从而激活细胞内的一系列信号通路,引起细胞状态的改变,参与了人体内几乎全部生理病理和药理过程[3]。其功能与多种疾病有关,如心血管类疾病、神经系统疾病、炎症疾病、代谢性疾病、癌症等。目前,超过40%的现代药物直接或间接靶向GPCRs[4]。
β肾上腺素受体(β-AR)是研究最深入的GPCRs之一[5-6]。其中,β2-AR在支气管平滑肌、心血管和肺部都有广泛表达,在调节心血管系统各项活动中扮演着至关重要的作用。目前市售β2-AR激动剂和拮抗剂较多,如沙丁胺醇(salbutamol)、特布他林(terbutaline)、沙美特罗(salmeterol)、福莫特罗(formoterol)和普萘洛尔(propranolol)等[7]。值得注意的是,上述β2-AR药物皆为正构配体,由于正构配体的高度保守性和受体基因序列的高度同源性,这些正构配体存在受体亚型选择性差、神经系统不良反应和禁忌症较多等问题[8]。与之相比,在空间和拓扑结构上,与正构结合位点不同的其他区域相结合的别构配体,因具有更高的受体亚型选择性而成为药物研发的热点[9-14]。目前,已报道的β2-AR别构调节剂有别构激动剂Cmpd6FA[15]和别构拮抗剂Cmpd15[16]。其中,Cmpd15的结构于2017年由本课题组与美国合作者共同报道(Chart 1)[17]。如图所示,Cmpd15可以由3个片段A~C偶联得到。其中片段A廉价易得,片段C的合成已由本课题组报道[18-19]。然而片段B对甲酰基取代的L-苯丙氨酸十分昂贵(7200元/g,安耐吉化学),不利于后续进行的Cmpd15衍生物的合成研究。
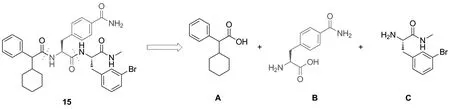
Chart 1
本文从二苯亚胺甘氨酸叔丁酯(2)和对甲酰胺苄溴(3)出发,经过不对称烷基化、水解、脱保护基和酰胺化等多步反应成功合成了Fmoc保护的片段B,即化合物12。通过在其羧酸端引入长链的聚乙二醇羧酸酯结构,成功测得了其ee值(97%)(Scheme 2),产物结构经1H NMR,13C NMR和HR-MS(ESI)确证。

Scheme 1
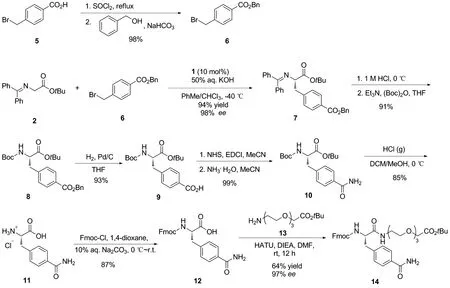
Scheme 2
1 实验部分
1.1 仪器与试剂
Autopol IV型旋光仪;Bruker 300/400 MHz型核磁共振仪(TMS为内标);Thermo Orbitrap Elite型质谱仪;Agilent 1260型高效液相色谱仪。
化合物1[20]、2[19]、3[21]和13[22]按文献方法合成;其余所用试剂均为分析纯。
1.2 合成
(1)4-溴甲基苯甲酸苄酯(6)的合成
在250 mL单口烧瓶中,加入4-溴甲基苯甲酸5(6.0 g, 27.9 mmol)和氯化亚砜(18 mL),回流反应3 h。冷却至室温,浓缩除去过量氯化亚砜,将苯甲醇(24 mL)和NaHCO3(7.0 g, 83.7 mmol)加入浓缩液中,搅拌5 h。抽滤,滤液浓缩后经硅胶柱层析[洗脱剂:V(石油醚)/V(乙酸乙酯)=100/1]纯化得白色固体6(8.3 g,收率98%);1H NMR(CDCl3, 300 MHz)δ: 8.04(d,J=8.4 Hz, 2H), 7.32~7.45(m, 7H), 5.36(s, 2H), 4.47(s, 2H);13C NMR(CDCl3, 75 MHz)δ: 165.9, 142.9, 136.0, 130.3, 130.1, 129.1, 128.7, 128.4, 128.3, 66.9, 32.3。
(2)(S)-4-(3-(叔丁氧基)-2-((二苯基亚甲基)氨基)-3-氧亚基)苯甲酸苄酯(7)的合成

(3)(S)-4-(3-(叔丁氧基)-2-((叔丁氧基羰基)氨基)-3-氧亚基)苯甲酸苄酯(8)的合成
将(S)-4-(3-(叔丁氧基)-2-((二苯基亚甲基)氨基)-3-氧亚基)苯甲酸苄酯7(1.0 g, 1.93 mmol)溶于四氢呋喃(17 mL)中,降温至0 ℃,滴加1 M HCl溶液(17 mL),滴毕,于0 ℃反应1 h。加入饱和碳酸氢钠溶液(10 mL)淬灭反应,用10% Na2CO3溶液调节pH至7~9,减压浓缩,残余物加入水(40 mL),用乙酸乙酯(3×40 mL)萃取,合并有机相,依次用饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩,粗产物溶于7 mL无水四氢呋喃中,于0 ℃滴加三乙胺(403 μL, 2.9 mmol)和二碳酸二叔丁酯(532 μL, 2.3 mmol),滴毕,反应1 h。用水(40 mL)淬灭反应,加入乙酸乙酯(3×40 mL)萃取,合并有机相,依次用饱和食盐水洗涤,无水硫酸钠干燥,浓缩,粗产物经硅胶柱层析[洗脱剂:V(石油醚)/V(乙酸乙酯)=20/1]纯化得白色固体8(800.4 mg, 91%);1H NMR(CDCl3, 400 MHz)δ: 8.00(d,J=8.1 Hz, 2H), 7.32~7.45(m, 5H), 7.25(d,J=7.2 Hz, 2H), 5.35(s, 2H), 5.04(d,J=4 Hz, 1H), 4.45~4.50(m, 1H), 3.06~3.17(m, 2H), 1.41(d,J=7.2 Hz, 18H);13C NMR(CDCl3, 100 MHz)δ: 170.7, 166.4, 155.1, 142.2, 136.1, 129.8, 129.7, 128.8, 128.7, 128.3, 128.3, 82.5, 79.9, 66.8, 54.7, 38.6, 28.4, 28.1。
(4)(S)-4-(3-(叔丁氧基)-2-((叔丁氧基羰基)氨基)-3-氧亚基)苯甲酸(9)的合成
将化合物8(250.0 mg, 0.6 mmol)溶于甲醇(3 mL)中,加入钯碳(37.5 mg, 15%),通入氢气,反应至终点。用硅藻土抽滤,滤饼浓缩得白色固体9(187.2 mg, 93%);1H NMR(DMSO-d6, 400 MHz)δ: 12.87(s, 1H), 7.85(d,J=8.0 Hz, 2H), 7.34~7.38(m, 2H), 7.24(d,J=8 Hz, 1H), 3.96~4.09(m, 1H), 3.01(dd,J=13.7 Hz, 5.6 Hz, 1H), 2.88~2.94(m, 1H), 1.29~1.34(m, 18H);13C NMR(DMSO-d6, 100 MHz)δ: 171.0, 167.3, 155.4, 143.1, 129.4, 129.2, 129.0, 80.5, 78.2, 55.6, 36.5, 28.1, 27.6。
(5)(S)-2-(((叔丁氧羰基)氨基)-3-(4-氨基甲酰基苯基)丙酸叔丁酯(10)的合成
氮气保护下,将9(150.0 mg, 0.4 mmol),N-羟基丁二酰亚胺NHS(61.2 mg, 0.5 mmol)和EDCI(103.3 mg, 0.5 mmol)溶于乙腈(5 mL)中,搅拌下反应12 h。浓缩,残余物加入水(10 mL),用二氯甲烷(3×10 mL)萃取,合并有机相,用无水硫酸钠干燥,浓缩,残余物用乙腈溶解,于0 ℃滴加氨水(1 mL),滴毕,搅拌1 h。浓缩,残余物加入水(10 mL),用二氯甲烷(3×10 mL)萃取,合并有机相,依次用饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析[洗脱剂:V(二氯甲烷)/V(甲醇)=20/1]纯化得白色固体10(148.4 mg, 99%);1H NMR(DMSO-d6, 400 MHz)δ: 7.9(s, 1H), 7.79(d,J=8.0 Hz, 2H), 7.30(d,J=7.8 Hz, 2H), 7.20(d,J=8.0 Hz, 1H), 4.01~4.07(m, 1H), 2.99(dd,J=13.7 Hz, 5.4 Hz, 1H), 2.86~2.91(m, 1H), 1.30~1.35(m, 18H);13C NMR(DMSO-d6, 100 MHz)δ: 171.1, 167.7, 155.4, 141.2, 132.4, 129.0, 127.4, 80.5, 78.2, 55.7, 36.3, 28.1, 27.6。
(6)(S)-4-甲酰胺基苯丙氨酸盐酸盐(11)的合成
将10(138.0 mg, 0.4 mmol)溶于二氯甲烷(2 mL)和甲醇(2 mL)中,通入氯化氢气体,反应至终点(TLC监测)。减压浓缩,残余物加入少量乙酸乙酯,抽滤,滤饼用乙酸乙酯洗涤得白色固体11(79.1 mg, 85%);1H NMR(DMSO-d6, 400 MHz)δ: 8.57(s, 3H), 7.89(d,J=8.1 Hz, 2H), 7.42(d,J=8.1 Hz, 2H), 4.19(s, 1H), 3.23(dd,J1=21.1 Hz,J2=14.8 Hz, 2H);13C NMR(DMSO-d6, 100 MHz)δ: 170.2, 140.3, 167.2, 129.8, 129.7, 129.5, 52.9, 35.5。
(7)(S)-9-芴基甲氧羰基-4-甲酰胺基苯丙氨酸(12)的合成
在25 mL的单口烧瓶中,加入化合物11(90.0 mg, 0.4 mmol),1,4-二氧六环和10%碳酸钠溶液,于0 ℃缓慢滴加Fmoc-Cl(95.2 mg, 0.4 mmol)的1,4-二氧六环(1 mL)溶液,滴毕,搅拌下于0 ℃反应4 h;升温至室温,反应16 h。用3 N盐酸溶液酸化反应体系至pH=1,加入水(20 mL),用乙酸乙酯(3×20 mL)萃取,合并有机相,依次用饱和食盐水洗涤,无水硫酸钠干燥,减压蒸馏,白色混合物加入少量乙酸乙酯,抽滤得白色粉末状固体12(138.3 mg,产率87%);1H NMR(DMSO-d6, 400 MHz)δ: 7.86(t,J=8.5 Hz, 4H), 7.77(d,J=8.5 Hz, 1H), 7.62(t,J=6.6 Hz, 2H), 7.38~7.42(m, 4H), 7.25~7.42(m, 2H), 4.14~4.24(m, 4H), 3.16(dd,J=13.7 Hz, 4.0 Hz, 1H), 2.91~2.97(m, 1H);13C NMR(DMSO-d6, 100 MHz)δ: 170.8, 169.8, 169.5, 155.9, 143.9, 141.4, 141.2, 132.0, 129.7, 127.9, 127.8, 127.2, 125.2, 120.1, 82.1, 70.7, 70.5, 70.4, 70.2, 69.8, 68.9, 67.0, 56.1, 47.2, 39.4, 39.1, 28.2; HR-MS(ESI)m/z: Calcd for C25H22N2O5{[M+Na]+}453.1421, found 453.1424。
(8)(S)-5-(4-氨基甲酰基苄基)-1-(9-芴基甲酯)-3,6-二氧亚基-2,10,13,16-四氧亚基-4,7-二氮杂十八烷-18-酸酯(14)的合成

为了实现片段B简洁高效的不对称合成,首先尝试使用对甲酰胺苄溴3与二苯亚胺甘氨酸叔丁酯2直接发生不对称烷基化反应构建片段B。但是遗憾的是,反应体系非常复杂,且产物很少(Scheme 1)。可能原因之一是化合物3在有机溶剂中的溶解性非常差。因此将对甲酰胺苄溴3中的酰胺替换为酯基后,与二苯亚胺甘氨酸叔丁酯2发生不对称烷基化反应。经过一系列筛选,最终选定了对甲酸苄酯取代的苄溴6作为反应底物。其中苄基可以通过催化氢化反应转化为相应的羧酸,之后通过氨基化反应得到片段B中的甲酰胺取代基。
从商业可得的对甲酸苄溴5出发,经过两步反应合成了所需的苄溴底物6。接下来苄溴6和二苯亚胺甘氨酸叔丁酯2在手性相转移催化剂1的作用下,发生不对称烷基化反应,以94%的产率和98%的ee得到了化合物7[20,23]。再经过亚胺水解和Boc保护得到化合物8。其中水解反应需要在0 ℃下进行,防止酯基同时被水解。化合物8发生催化氢化脱去苄基得到相应的羧酸9。化合物9经过胺化反应得到所需的含有对位甲酰胺取代基的化合物10。该步骤也可使用偶联试剂HATU/EDCI和NH4Cl进行酰胺化,产率94%。再用盐酸脱去Boc保护基得到片段B的盐酸盐化合物11,最后,将11进行Fmoc保护得到所需的12。由于12在有机溶剂中的溶解性很差,后处理可以用乙酸乙酯等有机溶剂进行多次洗涤,即可得到高纯度的12。
接下来,尝试测定此条路线合成的12的光学纯度。发现由于12在常见的有机溶剂中溶解度很低,仅能少量溶解在DMSO中,难以使用HPLC测定其ee值。因此,在其羧酸端引入了一条聚乙二醇羧酸长链,得到了化合物14,大大提高了其溶解度,最终测得化合物14的ee值为97%,与化合物7的ee值接近,证明了此条路线的可靠性。
从对甲酸苄溴出发,经过不对称烷基化反应,水解反应,胺化反应等10步反应合成了β2-AR别构拮抗剂Cmpd15的关键中间体片段B。此条合成路线产率高,对映选择性较好(97%ee)。该研究为Cmpd15及其类似物的合成提供了一条高效可靠的路线。
