GC-MS法测定果蔬汁中噻虫嗪的不确定度评定
2020-12-31梁馨予
梁馨予
(玉林市食品药品检验检测中心,广西 玉林 537000)
噻虫嗪,第二代新烟碱类杀虫剂,极性大[1-2],其作用机制与尼古丁相同,属于内吸性杀虫剂[3],是我国批准使用的农药品种。目前国家标准中针对食品中噻虫嗪相关的检测方法主要有液相色谱-串联质谱法(LC-MS/MS)[4-6],液相色谱-质谱法(LC-MS)[7-8],气相色谱-质谱法(GC-MS)[9]。目前农药残留的相关工作如市场监管,食品安全风险评估等,都是以初级农产品为对象展开的。而农药残留分析应考虑农药在加工过程中的残留。以果蔬榨汁为例,一般果蔬榨汁过程中,极性大,水溶性好的农药易进入果蔬汁中[10]。在2019年,我国农业农村部首次集中批准火龙果用药登记[11],其中包括了噻虫嗪成分。因此开展提高果蔬汁中农药残留检测的技术能力,对加强农药登记管理技术支撑尤为重要。以噻虫嗪为例,采用气相色谱-质谱法测定了果蔬汁中噻虫嗪的含量,并建立了不确定度评估。
1 实 验
1.1 仪器与试剂
TRACE 1310-TQS 8000 Evo气质联用仪,赛默飞世尔科技有限公司;噻虫嗪标准品(批号为G133046,含量以99.6%计,相对扩展不确定度U(k=2)为0.5%),DR.公司;丙酮(色谱纯,20 ℃的体积膨胀系数为1.37×10-3℃-1),默克股份两合公司;正己烷(色谱纯,20 ℃的体积膨胀系数为1.36×10-3℃-1),默克股份两合公司。
1.2 溶液制备
1.2.1 标准储备溶液(1 mg/mL)的制备
精密称取噻虫嗪标准品(DR.公司,批号为G133046,含量以99.6%计)约20 mg,置20 mL量瓶中,用丙酮溶解并定容至刻度,摇匀,4 ℃保存。
1.2.2 标准工作溶液(1 μg/mL)的制备
精密吸取噻虫嗪对照品(约1 mg/mL)0.10 mL于100 mL容量瓶中,正己烷定容至刻度,摇匀,4 ℃保存。
1.2.3 系列标准工作曲线溶液的制备
分别精密吸取噻虫嗪标准溶液(约1 μg/mL)0.05 mL、0.10 mL、0.20 mL、0.50 mL、1.00 mL于10 mL容量瓶中,正己烷定容至刻度,即得噻虫嗪系列标准工作曲线溶液。浓度分别约为5 ng/mL、10 ng/mL、20 ng/mL、50 ng/mL、100 ng/mL。
1.3 样品制备
称取10.0 g试样,于50 mL离心管中,精密加入1%醋酸乙腈溶液15.0 mL,涡旋振荡提取2 min,向离心管中加入1.5 g无水醋酸钠,再振荡2 min,再向离心管中加入6 g无水硫酸镁振荡2 min,4000 r/min 离心10 min,精密量取上清液10 mL置15 mL离心管中,40 ℃水浴中氮吹浓缩至近干,最后使样液体积为2.0 mL,混匀,用于气相色谱-质谱测定。
1.4 仪器条件
色谱柱:TG-5 MS毛细管柱(30 m×250 μm×0.25 μm);升温程序:初始温度80 ℃,保持1.0 min;25 ℃/min升至250 ℃,保持3 min;载气流速为1.2 mL/min;进样量:1 μL;不分流进样。电子轰击离子(EI)源,溶剂延迟时间为3 min,采集方式:选择离子监测(SRM)模式。
1.5 实验温度
控制在(20±5)℃内。
2 数学模型
噻虫嗪含量的表示式为
式中:X——试样中噻虫嗪的残留量,mg/kg
C——从标准工作曲线上得到的噻虫嗪浓度,ng/mL
V——样品定容体积,mL
m——试样质量,g
3 不确定来源分析
GC-MS法测定果蔬汁中噻虫嗪残留量的主要不确定度来源包括:标准储备液的配制、系列标准工作溶液的配制、样品的前处理、标准曲线拟合和测量重复性。
4 不确定度的分析与计算
4.1 标准储备液配制过程引入的不确定度urel(M)
4.1.1 电子天平称量引入的不确定度u1,rel(M)
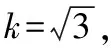
减量法称量,称量标准物质M=0.02230 g,由此产生的相对标准不确定度为:
4.1.2 标准物质的引入的不确定度u2,rel(M)
证书给出的相对扩展不确定度为U=0.5%,相对标准不确定度为:
4.1.3 标准储备液定容引入的不确定度u3,rel(M)
按照JJG196-2006《常用玻璃量器检定规程》[12],20 mL A 级量瓶容量的允许误差为±0.02 mL,按均匀分布,由此产生的不确定度为:
温度:实验环境温度为(20±5)℃,丙酮在20 ℃的体积膨胀系数为1.37×10-3℃-1,按均匀分布,由此产生的不确定度为:
以上两项引入的合成相对标准不确定度为:
4.1.4 合成由标准储备液配制过程引入的合成相对标准不确定度
4.2 标准系列工作曲线溶液配制过程引入的相对不确定度urel(V)
标准系列工作曲线溶液配制过程的不确定度主要由移液枪移取的体积、玻璃量器定容的体积、实验室的温度等引起。
4.2.1 标准工作溶液引入的不确定度u1,rel(V)
精密吸取噻虫嗪对照品(1.0 mg/mL)0.10 mL于100 mL容量瓶中,正己烷定容至刻度,配制后浓度约为1.0 μg/mL。
按照JJG 646-2006《移液器检定规程》[13],100 μL移液器吸取100 μL溶液允许误差为2.0%,按均匀分布,由此产生的不确定度为:
温度:实验环境温度为(20±5)℃,正己烷在20 ℃的体积膨胀系数为1.36×10-3℃-1,按均匀分布,由此产生的不确定度为:
由移液枪吸液引入的合成相对标准不确定度为:
JJG196-2006《常用玻璃量器检定规程》规定,100 mL A级量瓶容量的允许误差为±0.10 mL,按均匀分布,由此产生的不确定度为:
温度:实验环境温度为(20±5)℃,正己烷在20 ℃的体积膨胀系数为1.36×10-3℃-1,按均匀分布,由此产生的不确定度为:
由100 mL容量瓶引入的合成相对标准不确定度为:
综上所述,标准工作溶液配制过程引入的合成相对标准不确定度为:
4.2.2 标准系列曲线溶液配制引入的不确定度u2,rel(V)
稀释过程中由移液器带来的不确定度:标准曲线绘制时所使用移液器的量程有0.05 mL、0.10 mL、0.20 mL、0.50 mL、1.00 mL。
校准:按移液器检定规程规定,A 级容量允许误差分别为±3.0%、±2%、±1.5%、±1.0%、±1.0%则由体积校准引入的标准不确定度分别为:
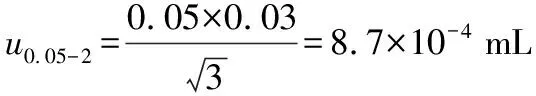
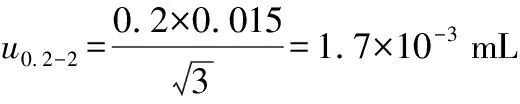
温度:实验环境温度为(20±5)℃,正己烷在20 ℃的体积膨胀系数为1.36×10-3℃-1,按均匀分布,由此产生的不确定度为:
JJG196-2006《常用玻璃量器检定规程》规定,10 mL A 级量瓶容量的允许误差为±0.020 mL,按均匀分布,由此产生的不确定度为:
温度:实验环境温度为(20±5)℃,正己烷在20 ℃的体积膨胀系数为1.36×10-3℃-1,按均匀分布,由此产生的不确定度为:
由10 mL容量瓶引入的合成相对标准不确定度为:
系列曲线溶液配制引入的相对标准不确定度分别为:
合成标准系列曲线溶液配制引入的相对标准不确定度分别为:
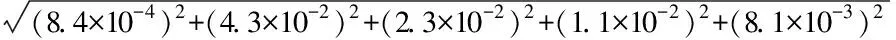
=5.1×10-2
所以,由标准溶液配制引入的相对标准不确定度为:
=5.1×10-2
4.3 样品前处理引入的相对不确定度Urel(R)
实验中严格按照标准方法执行,为了考察前处理过程中哪一个步骤对不确定度的贡献较大,因此对操作步骤进行逐步考量。
(1)15 mL A级单标线吸量管容量允许误差为±0.025 mL,按均匀分布,其标准不确定度为:
温度: 实验环境温度为(20±5)℃,乙腈在20 ℃的体积膨胀系数为1.37×10-3℃-1,按均匀分布,则由温度效应引入的标准不确定度为:
由以上两项引入的合成相对标准不确定度为:
(2)10 mL移液枪移取10 mL溶液允许误差为0.6%,按均匀分布,其标准不确定度为:
温度:实验环境温度为(20±5)℃,乙腈在20 ℃的体积膨胀系数为1.37×10-3℃-1,按均匀分布,则由温度效应引入的标准不确定度为:
由以上两项引入的合成相对标准不确定度为:
(3)1 mL移液枪移取2 mL溶液允许误差为1.0%,按均匀分布,其标准不确定度为:
温度:实验环境温度为(20±5)℃,正己烷在20 ℃的体积膨胀系数为1.36×10-3℃-1,按均匀分布,则由温度效应引入的标准不确定度为:
由以上两项引入的合成相对标准不确定度为:
(4)合成样品前处理过程引入的不确定度
4.4 标准曲线拟合和测量重复性引入的相对不确定度urel(C)
4.4.1 样品结果测量重复性的引入的不确定度u1,rel(C)
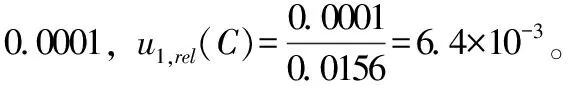
4.4.2 标准曲线拟合引入的不确定度u2,rel(C)
噻虫嗪的标准曲线方程与相关系数为:
Y=9.354×103X-1.424×104,r=0.9948
标曲峰面积:①32262、②72119、③186128、④728924、⑤993709;
标准溶液实际响应值:①4.972、②9.233、③21.421、④79.452、⑤107.760;
回归曲线所测理论值:①4.442、②11.105、③22.210、④74.030、⑤111.050;
根据《化学分析中不确定度的评估指南》[14],由标准曲线拟合引入不确定度由下式计算:
=1.7×10-5
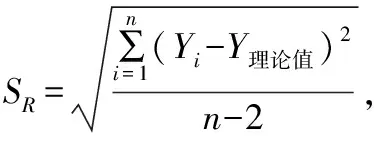
合成标准曲线拟合和测量重复性的不确定度urel(C)为:
=6.4×10-3
5 合成标准不确定度uc(X)
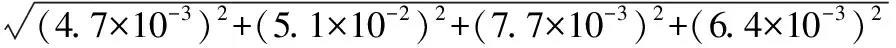
=0.052
由4.4.1得知:样品中噻虫嗪含量X=0.0156 mg/kg,则:
uc(X)=0.052×X=0.052×0.0156=0.0008 mg/kg
6 计算扩展不确定度U(X)
设定包含因子为2,则:
U(X)=uc(X)×2=0.0008×2=0.0016 mg/kg
7 结果与讨论
报告结果为(0.0156±0.0016) mg/kg,k=2。不确定度主要来源于标准溶液配制过程,因此在以后检测中注意合理使用移液器和容量瓶,首选A级容量瓶,其他几个分量引入的不确定度均比较小,所以得出的噻虫嗪残留量的值范围是合理的,测量结果是可靠的。
前处理过程中,发现对于非极性农药来说,乙腈和乙酸乙酯提取效率区别不大,但对于极性农药,乙腈提取效率要高。由于噻虫嗪是酸性农药,对碱比较敏感,在酸性条件下比较稳定[15],采用醋酸乙腈提取,发现提取效率比单纯乙腈更好。采用了萃取盐盐析,先加入无水醋酸钠缓冲盐使基质环境的pH保持在5~5.5之间,提高萃取效率,保证回收。溶剂消耗少,清洁环保,方法简单,操作步骤少,费用低,减少了人为因素对方法准确性的影响,适合简单基质果汁中噻虫嗪初筛检测。
近几年抽检工作经历,监督部门加强了对初级农产品农药残留的监测,但国家标准对加工品果蔬汁中噻虫嗪残留量判定是几乎空白的[16-18],这可能会导致农药残留随着加工品进入人体,对人的健康造成威胁。此外,2019年美国环保署取消了12项含噻虫胺、噻虫嗪成分新烟碱类农药登记[19],2019年加拿大将在2~3年内淘汰噻虫胺、吡虫啉、噻虫嗪某些用途[20],2018年法国禁止销售与使用五种新烟碱类杀虫剂(噻虫胺、吡虫啉、噻虫嗪、噻虫啉、啶虫脒),法国也成为欧盟首个为保护蜜蜂种群禁用新烟碱类杀虫剂的国家[21],2018年欧盟通过了禁止户外使用新烟碱农药的法令,全面禁止三种新烟碱类杀虫剂(吡虫啉、噻虫嗪和噻虫胺)户外应用,仅允许其施用于温室作物[22],2018-2020年日本加强对中国蒜苗、葱、胡萝卜、姜中噻虫嗪残留量的监控检查[23-26]。随着全球经济一体化的日益深入,在我国加强噻虫嗪等新烟碱类杀虫剂在加工品中的残留检测,提高检测技术能力,为农药登记管理和风险评估提供技术支撑,为登记使用、标准提升与国际接轨做好准备。
