卡那霉素A与腺嘌呤间氢键作用位点的研究
2020-12-29黄翠英姜笑楠
黄翠英, 姜笑楠, 王 萌
(辽宁师范大学 化学化工学院,辽宁 大连 116029)
氨基糖苷类抗生素(aminoglycoside antibiotics)是与RNA特异性结合的代表性药物小分子[1-2], 是广谱杀菌抗生素, 具有非常高的临床应用价值. 卡那霉素A是2-脱氧链霉胺类氨基糖苷类抗生素中4-6取代的代表性抗生素, 在临床上使用较多. 伴随着多重耐药性传染病的日益增加, 合成新的抗菌药物成为首要目标[3-4]. 为了实现这一目标, 深入理解氨基糖苷类抗生素与RNA的结合方式和结合强度十分必要. François等[5]通过对卡那霉素A、新霉素和新霉素B等药物分子与核苷酸配合物晶体结构的研究, 揭示了它们之间主要是氢键作用.
基于RNA与氨基糖苷类抗生素的特异性结合的诸多实验研究结果, 本文选取卡那霉素A为氨基糖苷类抗生素的代表, 研究其与腺嘌呤间的氢键相互作用, 探讨卡那霉素A与腺嘌呤间氢键作用的最佳位点.
1 研究对象与研究方法
1.1 研究对象
图1列出了卡那霉素A(KaA)和腺嘌呤(A)的结构、可能的相互作用位点和位点编号.本文利用“氨糖苷分子名称+作用位点编号-碱基名称+作用位点编号”的形式对氢键复合物进行系统命名. 如卡那霉素A(KaA)的1号位点与腺嘌呤(A)的1号位点形成氢键复合物命名为KaA1-A1.

图1 腺嘌呤(A)、卡那霉素A(KaA)和卡那霉素A核心部分(KA)的结构与可能的氢键作用位点Fig.1 Structures and hydrogen bonding sites of adenine (A), Kanamycin A (KaA), and the core part of Kanamycin A (KA)
通过分子动力学研究发现, 氨基糖苷类抗生素主要通过卡那霉素A核心部分(图1中的KA)与RNA碱基形成较强的分子间相互作用, 在分子的特异性识别和抗菌作用中具有重要地位[6-10]. 表1列出了KaA-A和KA-A体系的结构参数、在M06-2X-D3/aug-cc-pVDZ 方法下得到的气相相互作用能和计算所需的CPU时间. 如表1中键长和键角参数所示, 去掉附加环Ⅲ后对复合物的结构影响不大. 在相互作用能上, 绝对误差最大的一组复合物为KaA1-A2与KA1-A2, 它们的相互作用能分别为-36.66和-38.12 kJ·mol-1, 绝对误差为-1.46 kJ·mol-1, 相对误差δ为3.99%, 相互作用能计算结果相差不大. 在计算效率上, KaA-A与KA-A体系的CPU耗时差值最小为27 h, 最大差值可达到103 h左右. 由以上比较可知, 去掉环Ⅲ(附加环)对体系的结构和相互作用能影响不大, 但CPU耗时明显减少. 因此, 本文使用卡那霉素A核心部分(KA)代替卡那霉素A(KaA)研究它与腺嘌呤间的氢键作用.

表1 KaA和KA的1号、2号位点与腺嘌呤A的3个位点形成的氢键复合物的结构参数、使用M06-2X-D3/aug-cc-pVDZ方法计算得到的气相相互作用能和计算所需CPU时间
1.2 研究方法
在B3LYP/6-31+G(d,p)[11-12]水平下获得了卡那霉素A(KaA)、卡那霉素A核心部分(KA)与腺嘌呤形成氢键复合物的稳定结构和红外光谱数据. 在M06-2X-D3/aug-cc-pVDZ[13]水平下计算了卡那霉素A核心部分(KA)与腺嘌呤形成的9个氢键复合物的气相、蛋白相(ε0=5.6)和水相(ε0=78.3)的相互作用能(IEgas、IEprotein和IEwater), 进行了自然键轨道(NBO)分析和分子中原子(AIM)理论计算, 获得了复合物的氢键临界点电子密度和二阶作用稳定化能. 此外, 采用MP2/aug-cc-pVTZ方法获得了卡那霉素A核心部分(KA)与腺嘌呤分子在298 K时的质子化和去质子化反应焓变. 复合物的结构优化、频率计算、能量计算、NBO计算等采用Gaussian 09程序包[14]完成. AIM理论计算采用Multiwfn程序[15]完成.
2 结果与讨论
2.1 KA与腺嘌呤的氢键作用位点
图2是KA与腺嘌呤(A)形成的9个氢键复合物的稳定结构. 表2列出了9个氢键复合物中各氢键的类型、键长(r)、临界点电子密度(ρc)、总临界点电子密度(Σρc)、二阶稳定化能(Eij)、总二阶稳定化能(ΣEij), 以及各氢键复合物的气相、蛋白相和水相下的相互作用能(IEgas,IEprotein,IEwater).

图2 KA与腺嘌呤形成的9个氢键复合物的结构和氢键键长(nm)Fig.2 The optimized structures and the lengths of hydrogen bonds (nm) of 9 hydrogen-bonded complexes formed by the KA bonding with the adenine

表2 9个氢键复合物中各氢键的类型、键长、临界点电子密度、总临界点电子密度、二阶稳定化能、总二阶稳定化能, 以及各氢键复合物的气相、蛋白相和水相的相互作用能
如图2和表2所示,KA与腺嘌呤(A)形成的9个氢键复合物包含N—H…N、O—H…N和O…H—N三种类型的氢键.其中,KA1-A1、KA1-A2和KA1-A3三个复合物中均含有N—H…N和O…H—N类型的氢键,KA2-A1、KA2-A2、KA2-A3、KA3-A1、KA3-A2、和KA3-A3六个复合物中均含有O—H…N和O…H—N类型的氢键.由氢键键长可知,在3种不同类型的氢键中,O—H…N氢键键长最短,O…H—N次之,N—H…N最长;同种类型氢键键长越短,氢键强度越强,复合物越稳定.
由表2数据可知,KA的3个位点与腺嘌呤A1位点形成的复合物KA1-A1、KA2-A1和KA3-A1的气相相互作用能分别是-40.04、-64.33和-72.15 kJ·mol-1.由相互作用能的强弱可知:与腺嘌呤A1位点作用时, KA3与之形成的氢键复合物最稳定, KA2次之, KA1最不稳定. 在KA与腺嘌呤A3位点形成的3个氢键复合物中, 复合物的稳定性顺序为KA3-A3>KA2-A3>KA1-A3. 由此可得: 与腺嘌呤A3位点作用时, KA3与之作用形成的氢键复合物最稳定, KA2次之, KA1最不稳定. 同理, 比较卡那霉素KA的3个位点与腺嘌呤A2位点作用时, KA3位点与其形成的氢键复合物最稳定, KA2次之, KA1最不稳定. 由此可知, 与腺嘌呤同一位点相互作用时, KA3位点与腺嘌呤形成的氢键复合物的相互作用最强. 这与通过氢键键长推得的结果一致.
腺嘌呤的3个位点与KA1形成的KA1-A1、KA1-A2和KA1-A3氢键复合物, 均包含1条O…H—N型氢键和1条N—H…N型氢键. KA1-A1、KA1-A2和KA1-A3中O…H—N型氢键的键长分别为0.203、0.208和0.195 nm, N—H…N型氢键对应的氢键键长分别是0.223、0.224和0.217 nm. 同种类型氢键键长越短, 形成的复合物越稳定. 因此, 由氢键键长可得: 与KA1位点作用时, 腺嘌呤位点稳定性次序为A3>A1>A2.复合物KA1-A1、KA1-A2和KA1-A3的气相相互作用能分别是-40.04、-38.12 和-54.38 kJ·mol-1.由此可知, 与KA1位点作用时, 腺嘌呤A3位点与之作用最强, A1次之, A2最弱. 这与氢键键长所推得的稳定性次序一致. 分析KA2和KA3两个位点与腺嘌呤不同位点形成复合物的氢键键长和气相相互作用能, 可以得到相同结论:与KA同一位点作用时, 腺嘌呤A3位点形成的氢键复合物最稳定, A1次之, A2最不稳定.
2.2 AIM和NBO
由AIM和NBO分析得到的9个氢键复合物体系各氢键的临界点电子密度(ρc)、总临界点电子密度(Σρc)、二阶稳定化能(Eij)和总二阶稳定化能(ΣEij)的数据列在表2的第4~7列. 由表2中的数据可知, 对于N—H…N、O—H…N和O…H—N三种类型的氢键, O—H…N型氢键的临界点电子密度最高、二阶稳定化能最大, O…H—N型氢键次之, N—H…N型氢键的临界点电子密度最低、二阶稳定化能最小; 临界点电子密度越高, 二阶稳定化能越大, 复合物越稳定.
表2中, 腺嘌呤不同位点与KA1位点形成的氢键复合物KA1-A1、KA1-A2和KA1-A3的总临界点电子密度分别为0.036 2、0.033 6和0.041 2 a.u., 总二阶稳定化能分别为55.30、48.99和71.06 kJ·mol-1, 由此可知三者的稳定次序为KA1-A3>KA1-A1>KA1-A2. 同理, 由腺嘌呤的3个不同位点与KA同一位点形成的复合物的氢键总临界点电子密度和总二阶稳定化能可知, 腺嘌呤A3位点为最强作用位点, A1次之, A2最弱. 复合物稳定性的相对顺序与由气相相互作用能所得顺序一致.
KA不同位点与腺嘌呤A3位点作用时, 复合物KA3-A3的氢键总临界点电子密度和总二阶稳定化能分别为0.070 0 a.u.和177.91 kJ·mol-1; 复合物KA2-A3的氢键总临界点电子密度和总二阶稳定化能分别为0.062 2 a.u.和144.17 kJ·mol-1; 复合物KA1-A3的氢键总临界点电子密度和总二阶稳定化能分别为0.041 2 a.u.和71.06 kJ·mol-1. 由此可得: 在KA1-A3、KA2-A3和KA3-A3三个氢键复合物中, KA3-A3复合物的氢键总临界点电子密度最高, 总二阶稳定化能最大, 形成的氢键复合物最稳定. 同理, 比较KA不同位点与腺嘌呤A1、A2位点作用形成的复合物的氢键总临界点电子密度和总二阶稳定化能可得, 与腺嘌呤同一位点作用, KA3位点与之形成的复合物的氢键总临界点电子密度最高, 总二阶稳定化能最大, 形成的氢键复合物最稳定. 复合物稳定性的相对顺序与由气相相互作用能所得顺序一致.
2.3 溶剂的影响
碱基核苷存在于生物体内的细胞液中, 药物小分子进入生物体内与碱基相互作用, 因此,研究溶剂环境下复合物体系相互作用强弱的变化,对进一步理解氨基糖苷类抗生素与碱基间的相互作用有重要意义.
从表2中气相、蛋白相和水相3种环境下的相互作用能可知, 在不同环境中KA和腺嘌呤各个位点的强弱顺序仍保持一致. 例如, 腺嘌呤A1位点与KA的3个成氢键复合物KA1-A1、KA2-A1和KA3-A1的气相相互作用能分别为-40.04、-64.33和-72.15 kJ·mol-1, 蛋白环境中的相互作用能分别为-32.69、-52.38和-59.02 kJ·mol-1, 水环境中的相互作用能分别为-31.27、-48.57和-55.51 kJ·mol-1.无论是在气相、蛋白相还是水环境, 都可推得KA3位点与腺嘌呤形成的氢键复合物最稳定, KA2次之, KA1最不稳定.
由表2数据还可知, 随着介电常数的增大相互作用能减弱, 即复合物在气相中的相互作用能强于在蛋白环境下的相互作用能, 强于在水环境下的相互作用能. 例如, 复合物KA2-A2在气相、蛋白和水环境下的相互作用能分别为-62.16、-51.21和-47.36 kJ·mol-1, 相互作用能的强弱顺序为IEgas>IEprotein>IEwater.
2.4 质子化(去质子化)反应焓变
刘畅等[16]在研究核酸碱基与甘氨酸二肽间氢键作用的最佳位点时, 使用MP2/aug-cc-pVTZ//MP2/aug-cc-pVDZ方法计算了核酸碱基各个位点的质子化和去质子化反应焓变ΔH, 并利用其相对大小推测氢键复合物的稳定性. 碱基某位点的质子化反应焓变越负, 通过该位点形成的氢键复合物越稳定, 碱基某位点的去质子化反应焓变越小, 通过该位点形成的氢键复合物越稳定.
为了提高计算效率,尝试采用MP2/aug-cc-pVTZ//B3LYP/6-31+G(d,p)方法计算各位点的质子化和去质子化反应焓变, 计算结果如图3所示. 从计算结果可知, 本文方法(图3圆括号外数值)与刘畅等人[16]使用的方法(图3圆括号里数值)得到的质子化和去质子化反应焓变的数值相差不大, 因此本文使用的MP2/aug-cc-pVTZ//B3LYP/6-31+G(d,p)方法计算质子化和去质子化反应焓变是较为合理的.
图4为使用MP2/aug-cc-pVTZ//B3LYP/6-31+G(d,p)计算所得的KA的质子化和去质子化反应焓变. KA1(-198和388 kcal·mol-1)和KA2(-200和341 kcal·mol-1)相比, KA2的质子化焓变更负,KA2与KA3去质子化焓变更小, KA更倾向通过KA2位点与腺嘌呤形成氢键复合物. KA2与KA3位点的质子化反应焓变分别为-200和-203 kcal·mol-1, KA3位点的质子化反应焓变更负; KA2与KA3去质子化反应焓变分别为341和338 kcal·mol-1, KA3位点的去质子化反应焓变更小. 因此与KA2位点相比, KA3与腺嘌呤形成的氢键复合物更稳定. 因此, 由质子化反应和去质子化反应焓变的相对大小亦可推断, KA与腺嘌呤形成氢键复合物时, 最倾向使用KA3位点, 其次是KA2位点, 最不倾向使用KA1位点. 由此得到的卡那霉素A位点强弱性顺序与由相互作用能所得结论相一致.
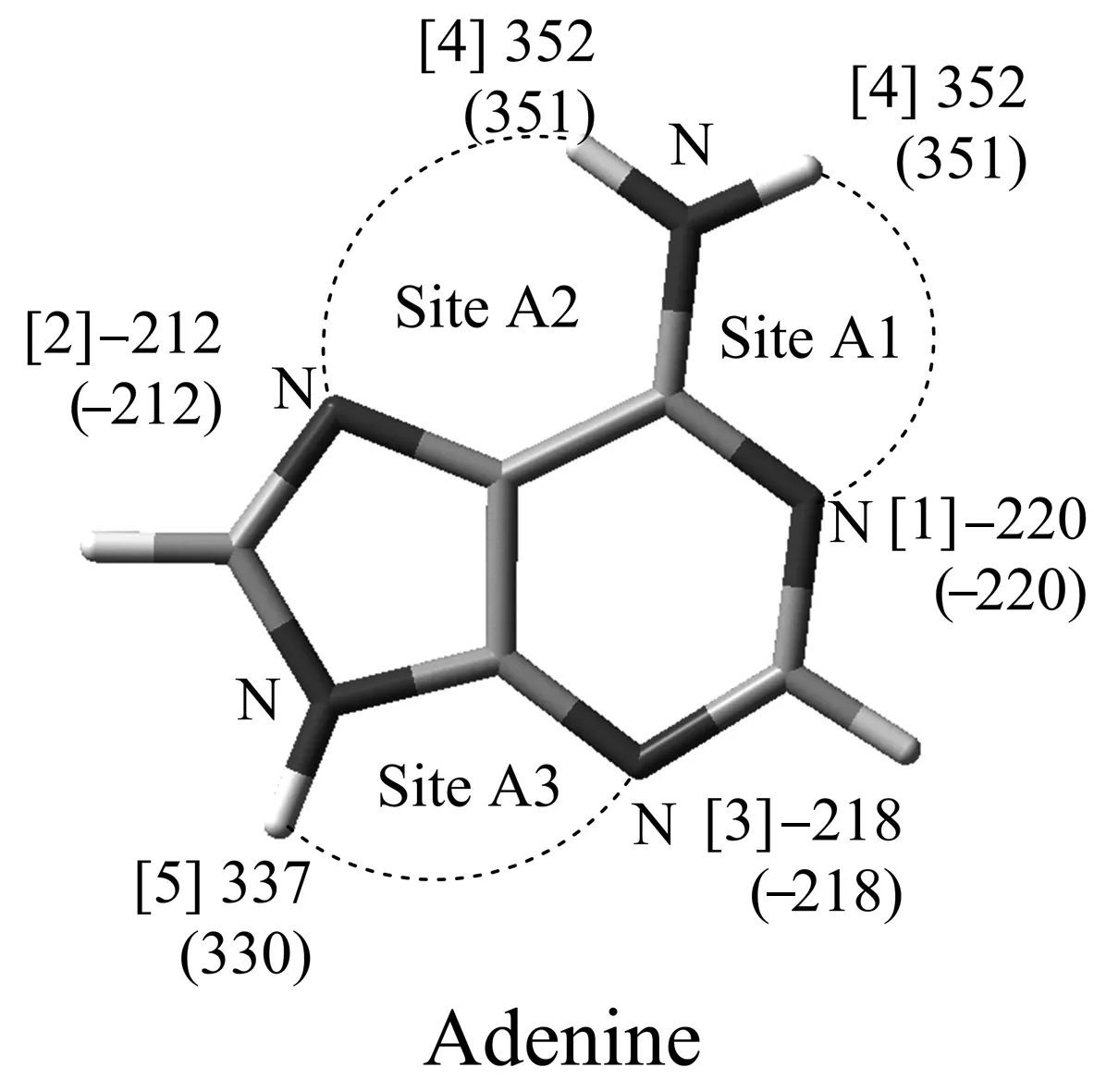
图3 腺嘌呤各氢键作用位点的质子化和去质子化反应焓变(单位:kcal·mol-1)Fig.3 The enthalpy values of the protonation and deprotonation reactions on the hydrogen bonding sites of the adenine

图4 卡那霉素 A 氢键作用位点的质子化与去质子化反应焓变(单位:kcal·mol-1)Fig.4 The enthalpy values of the protonation and deprotonation reactions on the hydrogen bonding sites of the Kanamycin A
3 结 论
通过研究可知,卡那霉素A更倾向于使用3号位点与腺嘌呤形成更稳定的氢键复合物, 腺嘌呤则倾向使用3号位点与卡那霉素A形成更稳定的氢键复合物. 由氢键复合物相互作用能计算得到的氢键复合物的稳定次序与卡那霉素A各位点的质子化和去质子化反应焓变推测得到的稳定次序一致. 希望本文研究结果能够对理解RNA碱基与卡那霉素A配体分子的相互作用提供理论帮助.
