PCR扩增技术联合CRISPR-Cas13a系统对MTB DNA检测方法的初步研究
2020-12-23于佳佳张旭霞张雨晴任卫聪姚丛李传友刘毅唐神结
于佳佳 张旭霞 张雨晴 任卫聪 姚丛 李传友 刘毅 唐神结
结核病是威胁人类生命健康的主要疾病,仍是全球十大死亡原因之一[1]。为实现2030年“终止结核病”的目标,早期、快速、准确的实验室诊断是提高结核病发现率和减少传播及发病的关键[2]。近年来,分子核酸检测已成为实验室诊断结核病广泛应用的重要手段[3],如GeneXpert MTB/RIF、线性探针、环介导等温扩增(LAMP)、荧光聚合酶链式反应(polymerase chain reaction,PCR)熔解曲线和基因芯片等检测方法,但因存在试剂贵、成本高、设备特殊、过程繁琐,以及检测时间较长、准确度和特异度较低等问题[4],难以在基层常规开展,迫切需要开发更快速、简便、价廉的高敏感度和高特异度的新型检测技术,以广泛应用于基层结核病的早发现、早治疗、早控制。
规律成簇间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)系统是存在于细菌和古细菌中的获得性免疫系统[5-6],其中Cas13a(CRISPR-associated 13a)蛋白具有RNA酶的活性[7],具有独特的靶向和切割机制,可在规律成簇间隔短回文重复序列RNA(CRISPR RNA, crRNA)的引导下识别单链核糖核酸 (single-stranded ribonucleic acid, ssRNA)[8],非特异性剪切非目标RNA[9-10]已被证明是一种高敏感度和高特异度的检测方法[11-13]。本研究将已高度成熟的PCR扩增技术与CRISPR-Cas13a检测技术相结合,通过对含MTB DNA的质粒模板、标准菌株H37Rv及6种非结核分枝杆菌进行敏感度及特异度分析,尝试建立一种更敏感、更准确、更快速、更简便的新型MTB核酸检测方法。
材料和方法
一、研究材料
1.菌株与质粒载体:戈登分枝杆菌(M.gordonae)、胞内分枝杆菌(M.intracellulare)、堪萨斯分枝杆菌(M.kansasii)、脓肿分枝杆菌(M.abscessus)、鸟分枝杆菌(M.avium)、偶发分枝杆菌(M.fortui-tum)等6种非结核分枝杆菌,以及大肠埃希菌E.coli和MTB H37Rv(ATCC 25618)菌株采用本实验室保存的菌株;质粒载体pMD19-Tsimple Vector 购自生工生物工程(上海)有限公司。
2.主要试剂:Cas13a蛋白由军事医学研究院微生物流行病研究所赠与;报告RNA试剂盒(RNase Alert v2, 30315288,Invitrogen公司)、鼠RNA酶抑制剂(Murine RNase inhibitor,M0314L,New England BioLabs公司)、T7 RNA聚合酶(T7 RNA Polymerase Mix,M0255A,New England BioLabs公司)、Ex Taq Version 2.0[TaKaRa, RR003,宝日医生物技术(北京)有限公司]、RNA纯化磁珠[Agencourt RNA Clean XP,Beckman Coulter,贝克曼库尔特商贸(中国)有限公司]、焦碳酸二乙酯处理的纯水 [DEPC treated water,等同于无核酸酶水(nuclease-free water)]、[pyrogen-free,46-2224,生工生物工程(上海)有限公司]、透明96孔板(Gene Protein,OS0329,Applied Biosystems公司)、封膜(Sealing Film,F601418-0001,BIO-RAD公司)等。
3.主要仪器:FLUOR-CHEM E凝胶成像系统(ProteinSimple公司)、ABI 7500荧光定量PCR仪(ABI公司)、DYY-3-4电泳仪(北京六一仪器厂)、Thermo Heraeus X1R台式高速冷冻离心机[赛默飞世尔科技(中国)有限公司]、Eppendorf Micro 21R冷冻型微量式离心机[赛默飞世尔科技(中国)有限公司]、哈东联BSC-1360II A2生物安全柜(浙江卫康检测科技有限责任公司)、HH.W21-Cr 420电热恒温水箱(北京长安永创科学仪器有限公司)、Axygen Maxygene-II梯度PCR仪(广州科适特科学仪器有限公司)、梅特勒ME204电子天平(上海恒平科学仪器有限公司)等。
二、研究方法
(一)构建模拟MTB质粒
将MTB保守序列IS6110片段插入pMDTM19-Tsimple Vector克隆载体,构建含待检测靶序列的质粒[基因片段由生工生物工程(上海)股份有限公司合成](图1)[14],用于设计含IS6110保守序列的模拟MTB质粒,以验证crRNA探针的有效性和特异性。
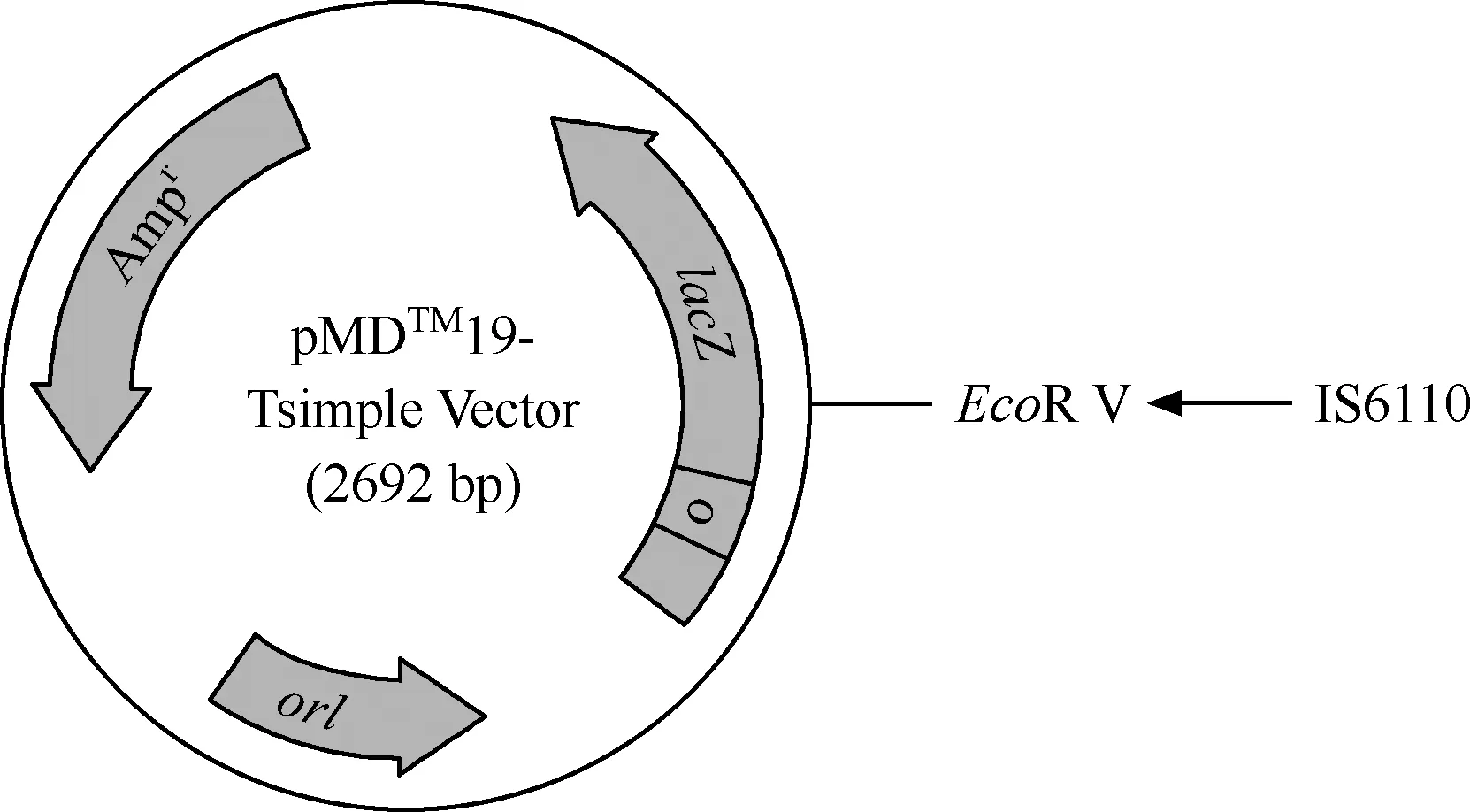
注 示意图来源于文献[14]。pMDTM19-Tsimple Vector:一种高效克隆PCR产物的专用载体;IS6110:MTB的保守序列。图中可见将MTB保守序列IS6110片段插入pMDTM19-Tsimple Vector克隆载体,构建含待检测靶序列的质粒图1 构建含待检测靶序列的质粒结构示意图
(二)设计MTB crRNA探针序列及引物
1.扩增模板的确立:利用MTB的保守序列IS6110,设计包含重复序列和检测靶点的规律成簇间隔短回文重复序列DNA(CRISPR DNA, crDNA)序列的扩增模板,所需序列及引物见表1。
2.crDNA的合成、产物回收:将设计的crDNA扩增模板及上下游引物加入至50 μl扩增体系中进行PCR扩增。回收扩增后的crDNA,用紫外分光光度计(nanodrop)测定不同浓度的相对荧光强度值,-80 ℃分装备用。
3.crRNA探针的转录及分离:将扩增后的含crDNA的扩增体系充分混匀后,37 ℃转录过夜(转录体系见表2),转录为实验所需的3条不同的crRNA,即IS6110-1crRNA、IS6110-2crRNA、IS6110-3crRNA,每条crRNA设置3个复孔,对构建成功的含有MTB DNA的质粒进行检测,对3个复孔的检测结果取均值(3个复孔每一次循环检测的相对荧光强度数值结果相加除以3得到每一循环的结果,共60个循环),比较3条不同crRNA信号的强弱,选择相对荧光强度值最高的1条crRNA用于后续实验检测。具体序列见表3。

表1 用于检测MTB DNA的crDNA序列及引物

表2 crDNA转录体系
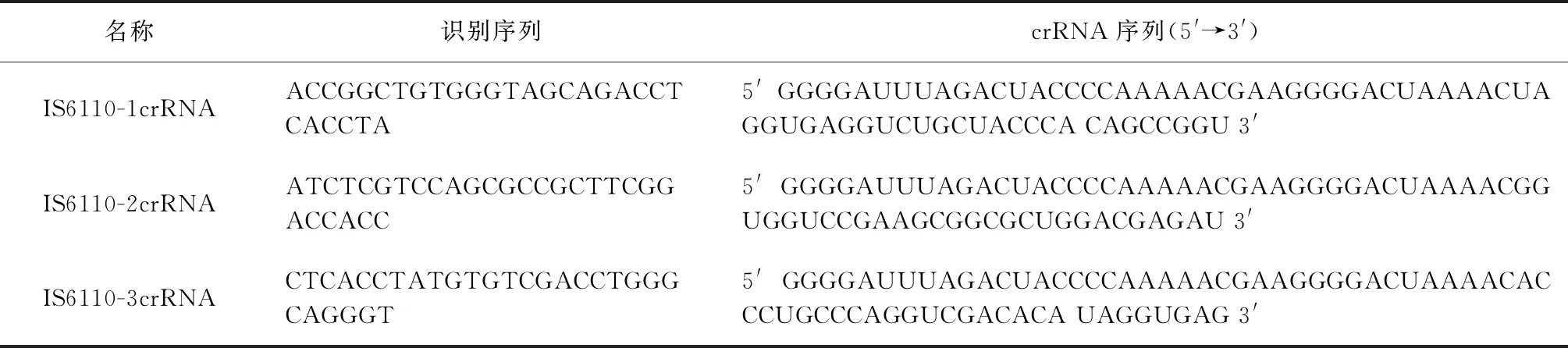
表3 用于检测MTB DNA的crRNA 序列
4.转录后crRNA的纯化:提前将磁珠振荡混匀,向转录产物中加入1.8倍体积的磁珠并涡旋30 s 以混匀磁珠和转录体系,室温静置3~5 min。再将反应体系置于磁力架上,静置5~10 min轻轻吸出体系中的液体,以分离磁珠。再向磁珠中加入70%的乙醇200 μl,室温孵育30 s,吸出乙醇;重复此过程清洗磁珠,共3次。室温晾干体系,去除体系中的乙醇,约10 min。加入50 μl nuclease-free water(阴性对照)溶解体系中的crRNA,涡旋30 s,吸出上清液,放入1.5 ml离心管中, nanodrop测定纯化得到的crRNA浓度的相对荧光强度值,-80 ℃分装备用。
(三)建立PCR-CRISPR检测方法和筛选crRNA序列
用特异性引物对待检测标本(梯度稀释的MTB质粒、H37Rv菌株及处理的非结核分枝杆菌)进行PCR扩增,再对得到的扩增序列进行琼脂糖凝胶电泳[阴性对照(NC)无条带时,说明扩增过程中无污染;将出现模糊条带视为其检测PCR产物敏感度的界值],检测扩增序列是否为实验所需的靶序列。将特异性引物扩增的靶序列(ssRNA,100 ng)、T7聚合酶(1 μl)、Cas13a蛋白(45 nmol/L)、crRNA(22.5 nmol/L)、NTP Buffer Mix(2 μl)、Murine RNase inhibitor(2 μl)、Background RNA(100 ng)等加入到反应体系中,构建不同待检测标本的PCR-CRISPR反应体系,并放入荧光定量PCR仪中检测。设置激发光波长为490 nm、37 ℃反应15 s,发射光波长为520 nm、37 ℃反应45 s,FAM通道检测相对荧光强度变化,共60个循环。仪器自动检测及报告RNA相对荧光强度变化情况(取均值)。
(四) PCR-CRISPR方法对含有MTB IS6110片段质粒检测的敏感度分析
将含有MTB IS6110片段的质粒进行梯度稀释(106、105、104、103、102、101、100拷贝/μl),每个模板取1 μl进行PCR 扩增,扩增后用T7聚合酶进行转录,收集纯化转录产物,用上述建立的PCR-CRISPR方法上机检测其相对荧光强度值,分析敏感度。
(五)PCR-CRISPR检测方法对MTB 标准菌株H37Rv的检测分析
将标准菌株H37Rv置于罗氏培养基37 ℃温箱内进行固体扩大培养3周,刮取菌株并研磨分散以备下一步实验用;测量处理后的菌株吸光度值(1个吸光度值相当于2×108CFU/ml)。对处理后的H37Rv菌株进行梯度稀释,得到浓度为 106、105、104、103、102、101、100拷贝/μl的模板,进行PCR扩增,扩增后用T7聚合酶进行转录,收集纯化转录产物,同样按上述 PCR-CRISPR方法检测相对荧光强度值,分析敏感度。
(六)PCR-CRISPR检测方法对MTB DNA特异度分析
对戈登分枝杆菌、胞内分枝杆菌、堪萨斯分枝杆菌、脓肿分枝杆菌、鸟分枝杆菌、偶发分枝杆菌等6种非结核分枝杆菌提取DNA后进行PCR扩增,使用PCR-CRISPR检测其产物的相对荧光强度值,分析该方法的特异度。
三、统计学处理
利用SPSS 24.0软件对数据进行统计学分析。采用GraphPad Prism 8.0.1(244) 绘制数据折线图和柱状图。非正态分布的数据采用中位数(四分位数)[M(Q1,Q3)]进行统计描述,并采用独立样本秩和检验(Kruskal-Wallis)进行多组数据的比较,以P<0.05为差异有统计学意义。如果多组比较差异有统计学意义,则进行不同检测组与对照组差异的两两比较,α′水平的调整采用Bonferroni校正,α′=0.05/7。
结 果
一、PCR-CRISPR检测含有MTB IS6110片段的模拟质粒
将模拟MTB质粒按照106→100拷贝/μl进行梯度稀释、PCR扩增,琼脂糖凝胶电泳检测扩增产物,显示无条带的阴性对照,以及清晰可见的103~106拷贝/μl的MTB DNA模板的电泳条带,且亮度随浓度的升高而增加(图2)。
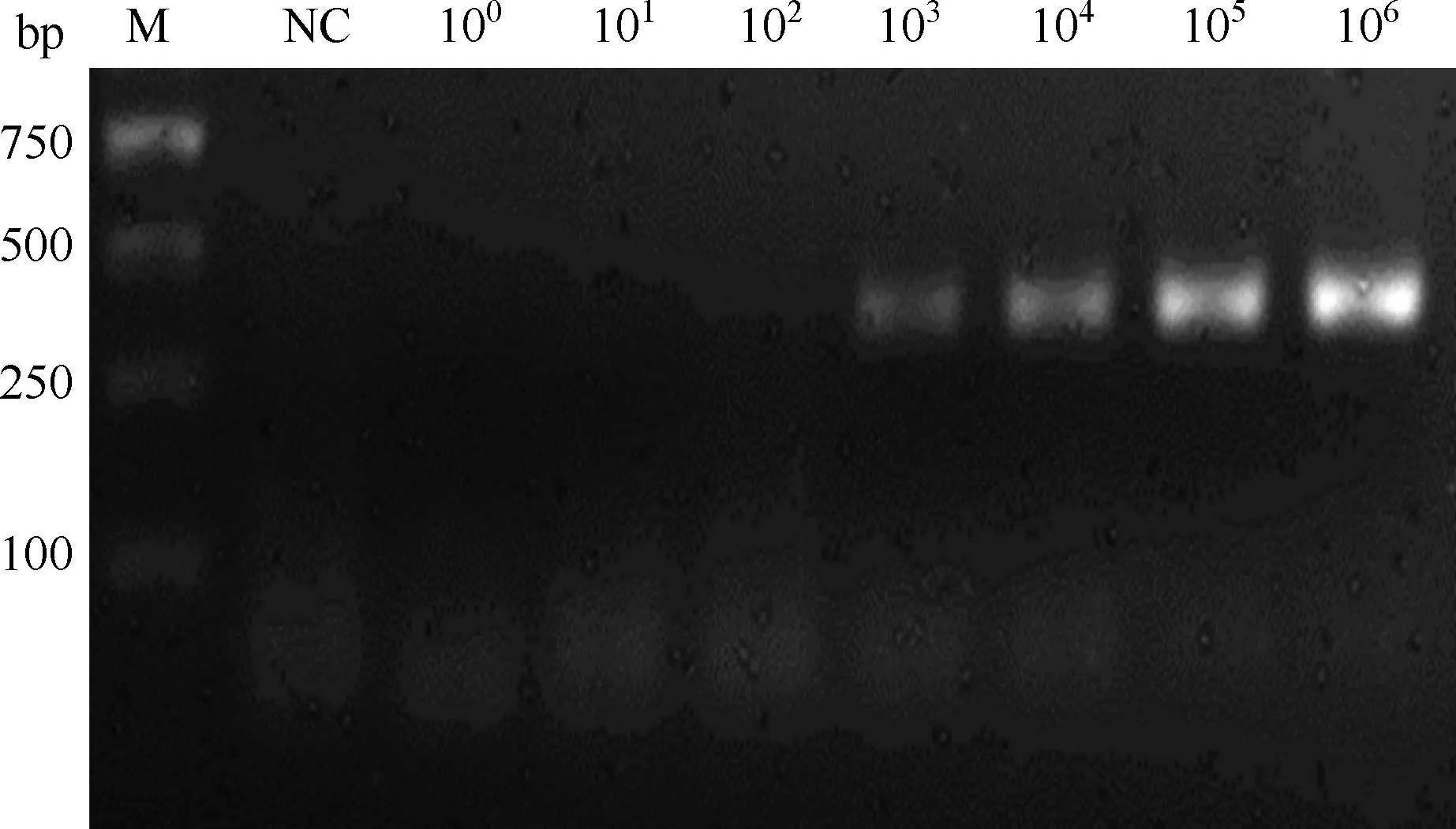
注 M:marker相对分子质量梯度;NC:阴性对照;100~106:分别为质粒标准品100~106拷贝/μl的PCR扩增产物电泳结果图2 琼脂糖凝胶电泳检测含有MTB IS6110片段的模拟质粒的扩增结果
二、IS6110-crRNA探针检测能力的比较
对MTB保守序列IS6110进行分析后,将3条不同的crRNA(即IS6110-1crRNA、IS6110-2crRNA、IS6110-3crRNA)与阴性对照分别对Cas13a 蛋白的特异性进行验证。结果显示,IS6110-3crRNA检测的相对荧光强度值从第1个循环的29 186.40 上升至第60个循环的46 184.80,M(Q1,Q3)为38 504.44(33 343.34,42 848.99),与阴性对照[38 794.06(38 509.60,39 076.07)]相近;而IS6110-1crRNA(从39 770.85上升至397 353.57)和IS6110-2crRNA(从46 709.64 上升至104 214.52)在识别目标核酸后均显示出较强的荧光信号[相对荧光强度值分别为197 680.64(98 364.94,304 271.25)和82 600.00(67 014.09,94 379.19)],分别约为阴性对照的10倍和3倍(图3)。因此,选择荧光信号最强的IS6110-1crRNA作为后续MTB DNA 检测的crRNA。
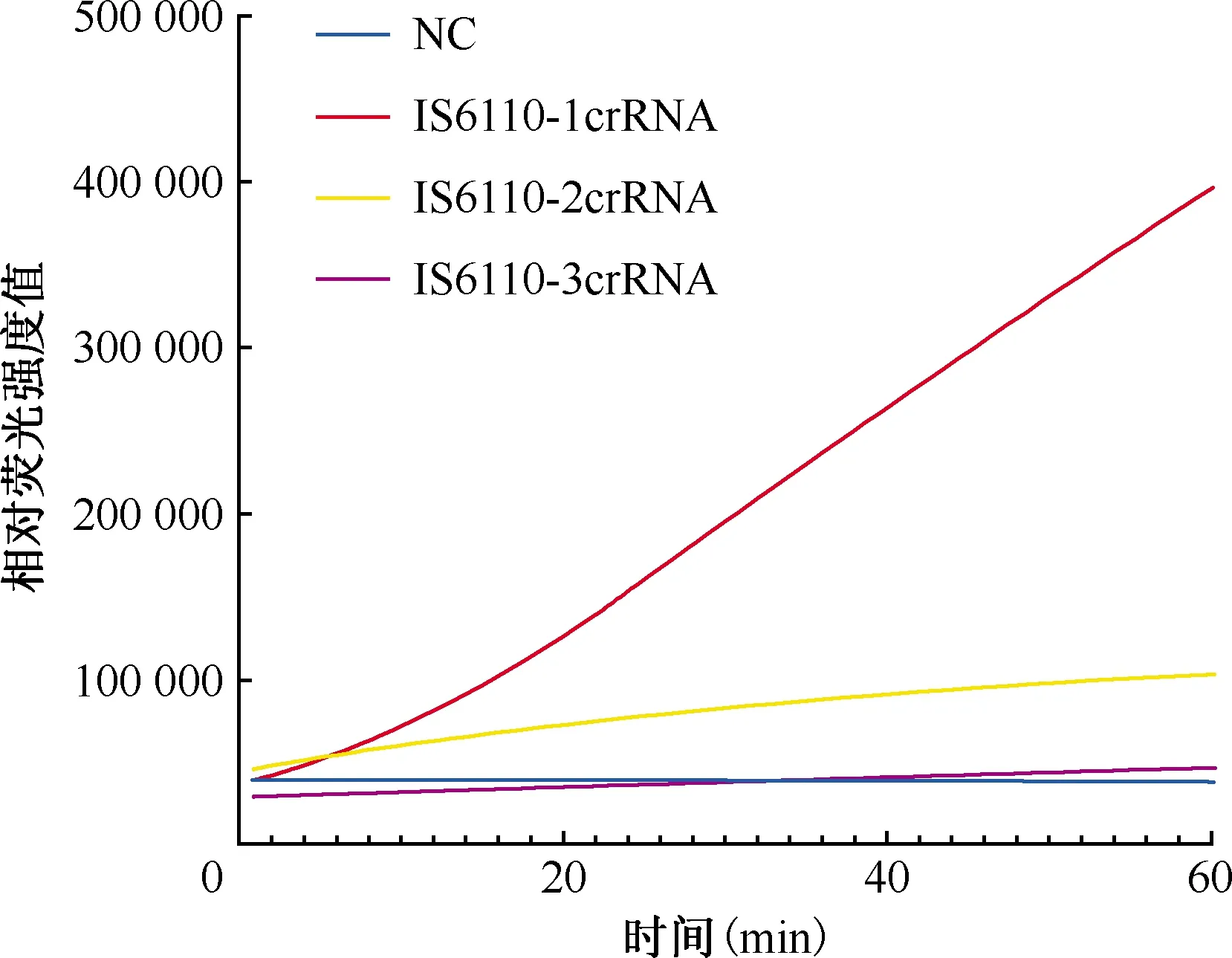
注 NC:阴性对照图3 3条不同crRNA探针检测能力的比较
三、 PCR-CRISPR方法对含有MTB IS6110片段质粒的检测
检测结果显示,106、105、104、103、102、101及100拷贝/μl质粒扩增产物的相对荧光强度值分别从第1个循环的22 203.66、22 087.28、19 740.01、22 632.79、30 592.57、28 511.19、21 262.39 上升至第60个循环的204 670.80、149 774.88、108 329.05、98 376.96、78 644.29、51 740.02、30 263.20,M(Q1,Q3)分别为113 272.44(68 467.39,159 309.84)、80 996.82(51 021.56,114 891.18)、61 000.13(39 817.42,83 940.77)、62 308.54(41 543.18,81 014.50)、50 154.71(36 952.00,64 654.68)、38 655.34(31 975.51,45 410.32)、28 739.11(25 216.65,32 359.91),与阴性对照[29 989.48(29 435.72,30 263.20)]总体比较差异有统计学意义(χ2=258.400,P<0.001)(图4,5),且发现随着模板稀释浓度的升高,不同浓度质粒模板的相对荧光强度增强;进一步进行两两比较,显示前6组质粒扩增产物的相对荧光强度值均明显高于阴性对照(Z=-8.503、-8.188、-6.986、-7.857、-9.369、-6.713,P值均<0.001),仅100拷贝/μl质粒扩增产物的数值与阴性对照差异无统计学意义(Z=-1.627,P=0.104)。
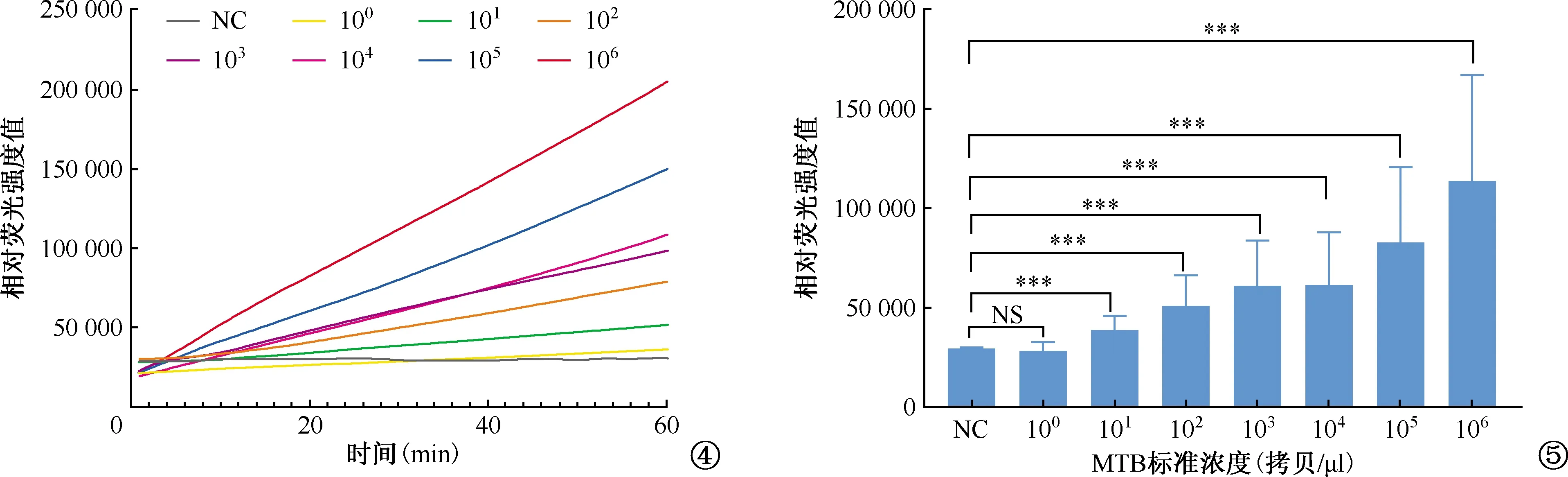
注 NC:阴性对照;100~106分别为质粒标准品100~106拷贝/μl的PCR扩增产物;NS:差异无统计学意义;***:P<0.001图4、5 分别为PCR-CRISPR检测MTB DNA质粒标准品敏感度的折线图和柱状图
四、 PCR-CRISPR检测标准菌株H37Rv的敏感度
利用MTB标准菌株H37Rv进一步评估PCR-CRISPR检测方法的敏感度。电泳结果可见无条带的阴性对照,以及清晰可见的104~106拷贝/μl的H37Rv标准品模板电泳条带,而103拷贝/μl时仅能观察到模糊的条带(图6)。
随后再利用梯度稀释的H37Rv标准品对PCR-

注 M:marker相对分子质量梯度;NC:阴性对照;100~106分别为标准菌株H37Rv稀释浓度100~106拷贝/μl的PCR扩增产物电泳结果图6 琼脂糖凝胶电泳检测H37Rv的扩增结果

注 NC:阴性对照;100~106分别为标准菌株H37Rv稀释浓度100~106拷贝/μl的PCR扩增产物;***:P<0.001图7、8 分别为PCR-CRISPR检测标准菌株H37Rv敏感度的折线图和柱状图
CRISPR检测方法进行评估。106、105、104、103、102、101及100拷贝/μl的H37Rv扩增产物的相对荧光强度值变化分别从第1个循环的26 351.51、24 139.54、14 191.19、16 998.90、18 593.55、17 513.12、17 498.48 上升至第60个循环的89 488.21、82 411.36、63 195.01、65 233.19、41 911.39、45 363.23、18 016.47,M(Q1,Q3)分别为68 115.15(49 224.36,80 863.30)、63 205.56(45 458.62,74 907.73)、41 007.78(25 503.90,53 859.83)、37 679.07(23 630.95,52 455.66)、27 503.72(21 049.85,34 784.11)、27 571.79(20 219.58,36 395.17)、17 691.50(17 612.36,17 793.29),与阴性对照[13 725.83(13 652.43,13 804.95)]总体比较差异有统计学意义(χ2=345.600,P<0.001);且发现随着H37Rv模板浓度的升高,不同浓度H37Rv模板的荧光强度增强(图7,8)。进一步进行两两比较,显示上述7组的相对荧光强度值均明显高于阴性对照(Z值均=-9.448;P值均<0.001)。
五、PCR-CRISPR检测MTB DNA的特异度
利用6种非结核分枝杆菌评估PCR-CRISPR检测方法的特异度。电泳结果可见无条带的阴性对照、6种非结核分枝杆菌电泳结果,以及清晰可见的106拷贝/μl的MTB DNA质粒的电泳条带(图9)。

注 M:marker相对分子质量梯度;NC:阴性对照;106:为质粒标准品106拷贝/μl的PCR扩增产物电泳结果;1:戈登分枝杆菌;2:胞内分枝杆菌;3:堪萨斯分枝杆菌;4:脓肿分枝杆菌;5:鸟分枝杆菌;6:偶发分枝杆菌图9 琼脂糖凝胶电泳检测6种非结核分枝杆菌及106拷贝的MTB DNA质粒的扩增结果

注 NC:阴性对照;106:质粒标准品106拷贝/μl的PCR扩增产物;***:P<0.001图10、11 分别为PCR-CRISPR检测6种非结核分枝杆菌及106拷贝MTB DNA质粒特异度的折线图和柱状图
随后利用阴性对照、6种非结核分枝杆菌及106拷贝/μl MTB DNA质粒标准品对PCR-CRISPR方法进行检测,结果可见,阴性对照 [37 635.57(37 168.74,38 199.20)]、戈登分枝杆菌[39 351.83(38 903.70,39 769.53)]、胞内分枝杆菌[39 191.30(39 018.51,39 434.95)]、堪萨斯分枝杆菌[25 172.20(24 586.95,26 046.45)]、脓肿分枝杆菌[37 328.03(36 959.01,37 546.78)]、鸟分枝杆菌[37 942.29(37 455.63,38 401.13)]、偶发分枝杆菌[29 491.19(29 148.63,30 058.62)]等6种非结核分枝杆菌的相对荧光强度值与106拷贝/μl的MTB DNA质粒的数值[从第1个循环的46 859.64上升至第60个循环的124 344.52,M(Q1,Q3)为89 204.07(66 253.60,108 819.13)]总体比较差异有统计学意义(χ2=441.900,P<0.001);进一步进行两两比较,显示7组相对荧光强度值均明显高于阴性对照(Z值均=-9.448,P值均<0.001)(图10,11)。特异度良好。
讨 论
结核病是引起人类死亡的重大传染性疾病,我国目前结核病疫情仍然十分严峻[1]。将PCR扩增和CRISPR-Cas13a检测技术相结合建立一种高敏感度、高特异度的核酸检测方法,对结核病的早期及准确诊断具有积极意义,为开发体外核酸检测的高敏感度诊断工具提供了新思路,同时也为PCR-CRISPR检测技术应用于临床标本检测提供了重要的理论依据。
CRISPR检测方法在病毒检测中的开发较多,在细菌中开发较少。目前,Gootenberg等[13]建立了高敏感度和高特异度的SHERLOCK核酸检测方法,并将之应用于生物样本(血液或尿液)中多种病毒的检测;East-Seletsky等[12]利用CRISPR-Cas13a系统对10 pmol/L 的RNA分子进行检测。Ai等[15]开发了一种基于CRISPR-Cas12a的MTB快速检测方法——CRISPR-MTB,并对179例患者进行回顾性队列研究,发现CRISPR-MTB(79%)相较于培养(33%)和GeneXpert MTB/RIF(66%)显示出更高的敏感度;且在不同类型的样本中,CRISPR-MTB相较于培养和GeneXpert MTB/RIF具有更强的整体诊断性能,为肺结核及肺外结核新的诊断技术提供了巨大的潜力;其中Cas12a可将识别的双链DNA (double-stranded DNA,dsDNA)作为激活剂,并可切割单链DNA (single-stranded DNA,ssDNA);但与靶向DNA的CRISPR相关酶(如Cas9a和Cas12a)不同,Cas13a能够在切割靶RNA之后保持活性,并且可在一系列被称作“附带切割(collateral cleavage)”的作用中继续切割其他的非靶向RNA。另外,CRISPR-Cas13a检测技术也在寨卡和登革热[16]、EBV[17]、埃博拉[18]、SARS-CoV-2[19]等病毒中显示出较高的敏感度,提示Cas13a能够被用来实现较高敏感度的检测,可广泛应用于科学和临床医学领域。
本研究利用PCR技术稳定性好、实用性强及高效扩增靶序列的特点,建立了基于PCR和CRISPR-Cas13a系统的MTB DNA检测方法——PCR-CRISPR。本检测方法首先设计含IS6110保守序列的质粒,将MTB保守序列IS6110片段插入pMDTM19-Tsimple Vector克隆载体,构建含待检测靶序列质粒,随后筛选出1条可用于MTB检测的crRNA,以模拟MTB质粒验证crRNA探针的有效性和特异性。然后利用PCR-CRISPR检测方法应用于MTB扩增、转录、crRNA 识别和 Cas13a 剪切等一系列特定过程,将扩增后的目标核酸进一步放大。该检测技术所应用的Cas13a蛋白是RNA引导下的靶向单蛋白效应器,具有独特的RNA靶向机制,是将独特的crRNA与靶点ssRNA相结合以引起Cas13a 构象变化,从而激活Cas13a强大的“平行切割效应”,使得Cas13a高效地剪切体系中的报告RNA,释放荧光信号,通过检测报告RNA释放的相对荧光强度来判断体系中是否存在靶序列。但需要注意的是,只有扩增产物转录为相应的ssRNA时,后续才能进行相应的crRNA识别及Cas13a激活等过程。基于此,即使对于低浓度的MTB DNA,也可检测到明显高于阴性对照的荧光信号,检测出体系中目标核酸的存在。
本研究显示,PCR-CRISPR检测MTB模拟质粒或H37Rv菌液的相对荧光强度值与阴性对照,以及阴性对照和6种非结核分枝杆菌与106拷贝/μl质粒标准品的相对荧光强度值的差异均有统计学意义,提示该方法敏感度及特异度良好,可检测待测样本是否感染或含有MTB。本研究利用PCR-CRISPR检测方法检测不同浓度质粒模板和标准菌株H37Rv的敏感度,结果显示在拷贝数101拷贝/μl和100拷贝/μl就可以达到较好的敏感度。再通过检测6种非结核分枝杆菌评估PCR-CRISPR检测方法的特异度,结果显示PCR-CRISPR同时具有较好的特异度。本研究建立了新的检测方法和系统,并进一步验证了该检测方法在检测MTB中的可行性和可靠性,下一步将利用PCR-CRISPR检测方法对临床菌株或患者进行验证评估。基于等温扩增技术开发的PCR-CRISPR检测技术,因其核酸检测功能具有可扩展性和高度复用能力,可将诊断和监测工作从样本的定向检测转移到大样本集的综合检测,有望在全球大部分地区实施,实现对感染性疾病的快速诊断。
综上所述,基于PCR扩增和CRISPR-Cas13a系统,本研究首次建立了针对MTB DNA的PCR-CRISPR核酸检测技术,具有敏感度高、特异度强、稳定性好及实用性强等特点,且成本更低,有望在临床检测实践中广泛推广。
志谢本研究统计学处理由首都医科大学附属北京胸科医院流行病统计学室康万里研究员指导!
