脂联素基因修饰对大鼠骨髓来源的内皮祖细胞活性的影响及机制探讨
2020-12-21陈丽芳戴颖仪林宋斌卢海克刘新通
陈丽芳,戴颖仪,林宋斌,卢海克,刘新通
(1.福建省莆田市涵江区平民医院超声科,福建 莆田 351115;2.南方医科大学第二临床医学院,广东 广州 510280;3.广东省第二人民医院神经内科,广东 广州 510317)
内皮祖细胞(endothelial progenitor cells,EPC)是血管内皮的前体细胞,来源于骨髓或者外周血细胞,参与新生血管的形成[1]和损伤血管修复[2]。EPC作为重要的内源性血管修复因素,在机体循环中其正常数量和功能的维持被认为与血管损伤性疾病密切相关[3,4]。循环内EPC在组织缺血损伤后可内源性动员,归巢于缺血部位,并且分化为成熟的内皮细胞参与血管新生[5],这为缺血性疾病治疗提供了新思路。许多研究表明EPC移植对缺血性脑卒中有积极的治疗作用[6,7]。然而在病理条件下,EPC的迁移、归巢和分化能力受损[8,9]。基因修饰是一种有潜力的提高EPCs迁移、增殖和分化能力的方法。许多研究表明移植基因修饰的EPC能够有效修复血管损伤[10,11]和缺血造成的损伤[12-14]。
脂联素(adiponectin,APN)是大量存在于血液中的主要由脂肪细胞分泌的细胞因子,除了具有改善机体血脂、血糖代谢的作用外,还具有抑制动脉粥样硬化、促进缺血组织局部血管新生等作用[15,16 ]。研究表明,APN促进EPC的增殖、迁移、分化和形成管腔样结构[17,18],而且与EPC治疗的大脑中动脉闭塞(middle cerebral artery occlusion,MCAO)大鼠相比,APN基因修饰EPC治疗组的行为功能、梗死面积百分比、微血管密度和细胞凋亡率的改善更明显[13]。虽然有研究表明球形脂联素通过恢复内皮一氧化氮合酶(endothelial nitric oxide synthase,eNOS)的活性改善高糖抑制的内皮祖细胞活性[19],但是在正常及其它病理条件下,APN改善EPC活性的机制尚不明确。在培养的大鼠心肌细胞中,脂联素治疗刺激血管内皮生长因子(vascular endothelial growth factor,VEGF)的生成[20],而VEGF通过诱导连接蛋白43(connexin 43,Cx43)表达促进EPC分化[21]。因此推测APN通过促进VEGF表达和eNOS活化增强EPC的功能。eNOS的活性受多个位点磷酸化调控,研究最为透彻的两个位点是激活位点丝氨酸残基1177(Ser1177)和抑制位点苏氨酸残基495(Thr495)[22]。基于上述分析,本研究旨在探索APN基因修饰对EPC的活性以及EPC中VEGF表达水平、eNOS活性的影响。
1 材料与方法
1.1 实验材料
3-4周龄健康雄性SD大鼠购自广东省医学实验动物中心,体重约90 g。293T细胞购自广州辉园苑医药科技有限公司(C14126)。pLKIRG载体(广州辉园苑医药科技有限公司,P16012),Ficoll-Paque PLUS(GE Healthcare,17-5442-02),DMEM培养基(Gibco,SH30022-01B),FBS(Hyclone,1027-106),胰蛋白酶(Sigma,T1426-250MG),TRIzol试剂(Invitrogen,15596026),qPCR Lentivirus Titration Kit(abm,Lv900),SYBR GREEN(TOYOBO,QPK-201),RIPA裂解液(碧云天,P0013B),Adiponectin小鼠单克隆抗体抗体(abcam,ab22554),VEGF小鼠源单克隆抗体(ABclonal,A17877),磷酸化VEGFR 2(Tyr1175)兔源多克隆抗体(AP0382,ABclonal),eNOS兔源多克隆抗体(A1548,ABclonal),磷酸化eNOS (Thr1177)兔源多克隆抗(北京博奥森生物技术有限公司,bs-3447R),GAPDH兔多克隆抗体(proteintech,10494-1-AP),CCK8试剂(碧云天,C0039)、结晶紫(BIOSHARP,Amresco 0528)。
1.2 大鼠EPCs的分离与培养
异氟烷麻醉处死SD大鼠后,迅速在无菌条件下分离股骨和胫骨,剪除骨干两端骨骺,显露髓腔,用1 mL培养基(无血清)冲出骨髓于离心管中,吹打制成细胞悬液。600×g离心5 min,弃去上清液,采用Ficoll-Paque PLUS溶液密度梯度离心法分离单个核细胞,PBS 洗涤后用1 mL EGM-2 BulletKit培养基重悬单个核细胞,接种到10 cm培养皿中,置于37℃、5%CO2、90%湿度的培养箱中定向诱导培养。72 h后,用培养基清洗未粘连的细胞,贴壁细胞用新鲜EGM-2 BulletKit培养基进一步培养,每隔2天半量换培养基。用显微镜观察EPC形态。诱导培养14 d后,细胞达到80%-90%融合时,用胰蛋白酶消化后按照1:3比例传代。
1.3 APN慢病毒真核表达载体克隆构建
使用 TRIzol 法提取大鼠EPC的总RNA,逆转录出cDNA。以cDNA为模板,用APN-F和APN-R引物进行PCR,扩增APN的编码序列,引物的序列见表1。PCR扩增产物和pLKIRG载体用NheⅠ、 XmaⅠ切割,切胶回收后,将APN的编码序列连接到慢病毒真核表达空载体pLKIRG中。APN慢病毒真核表达载体用NheⅠ、XmaⅠ双酶切检测,并且送华大基因公司进行测序验证。
1.4 慢病毒包装及病毒滴度检测
提前1 d把293T细胞接种到10 cm培养皿,细胞融合度达到80%左右时,将pLKIRG载体、APN慢病毒真核表达载体分别与慢病毒包装载体PPACK-H1-GAG、PPACK-HI -REV、PVSV-G按照3:1:1:1的比例转染293T细胞,转染后24、48 h时分别收集细胞培养液, 4℃,82 700×g超速离心2 h浓缩培养液中的慢病毒颗粒,按照qPCR Lentivirus Titration Kit的操作手册检测病毒滴度。
1.5 慢病毒感染细胞及筛选稳定细胞株
EPC细胞提前一天接种到6孔板中,细胞融合度达到60%左右时,用APN基因重组慢病毒及对照慢病毒分别感染EPC细胞,感染10 h后换为正常培养基,慢病毒感染48 h后用0.1 μg/μl嘌呤霉素筛选3 d,然后收集细胞。
1.6 Real-time PCR 检测
使用 TRIzol 法提取细胞总RNA,逆转录出cDNA。用SYBR GREEN进行Real-time PCR检测,选择GAPDH基因作为内参,使用的引物的序列见表1。根据荧光定量PCR结果的CT值计算试验结果,目的基因的相对表达水平=2-△△Ct。

表1 引物的序列
1.7 Western blot检测
PBS漂洗各组EPC,加入冰预冷的RIPA裂解液裂解细胞,12 000×g离心10 min,收集上清液,取5 μL上清液测定蛋白浓度。每组20 μL样品用SDS-聚丙烯酰胺凝胶电泳分离,转移至醋酸纤维素薄膜上,室温下用5% BSA封闭3 h后加入相应的一抗,4℃孵育过夜, TBST洗膜3次,室温下用二抗孵育2 h,TBST洗膜3次,ECL显色,用成像系统拍照。选择GAPDH作为内参。
1.8 CCK8检测细胞增殖
消化生长状态良好的各组细胞,制备25 000个/mL的细胞悬液,接种于96 孔板中,每孔接种200 μL,每组5个孔。将培养板放在37℃,5% CO2培养箱中预培养4、24、48 h和72 h。向每孔加入20 μL CCK8溶液,将培养板在培养箱内孵育2 h,用酶标仪测量450 nm波长的吸光度。
1.9 Transwell检测细胞迁移
消化生长状态良好的各组细胞,制备细胞悬液,接种5×104细胞于上室,下室补充500 μL完全培养基,将培养板放在37℃、5% CO2培养箱中培养24 h。取出Transwell小室,用PBS清洗细胞,然后用棉签擦去上室内的细胞,将贴附于微孔膜下层的细胞用多聚甲醛固定20 min,结晶紫染色后用刀片将微孔膜割下,放置在载玻片上,滴加中性树胶,盖上盖玻片,显微镜下观察,400倍视野下随机选取5个视野拍照、计数,取平均数。
1.10 统计学方法
应用SPSS 21.0 统计学软件进行分析,计量资料以均数±标准差表示,两组比较采用两独立样本t检验,P<0.05为差异有统计学意义。
2 结果
2.1 大鼠EPC的培养及鉴定
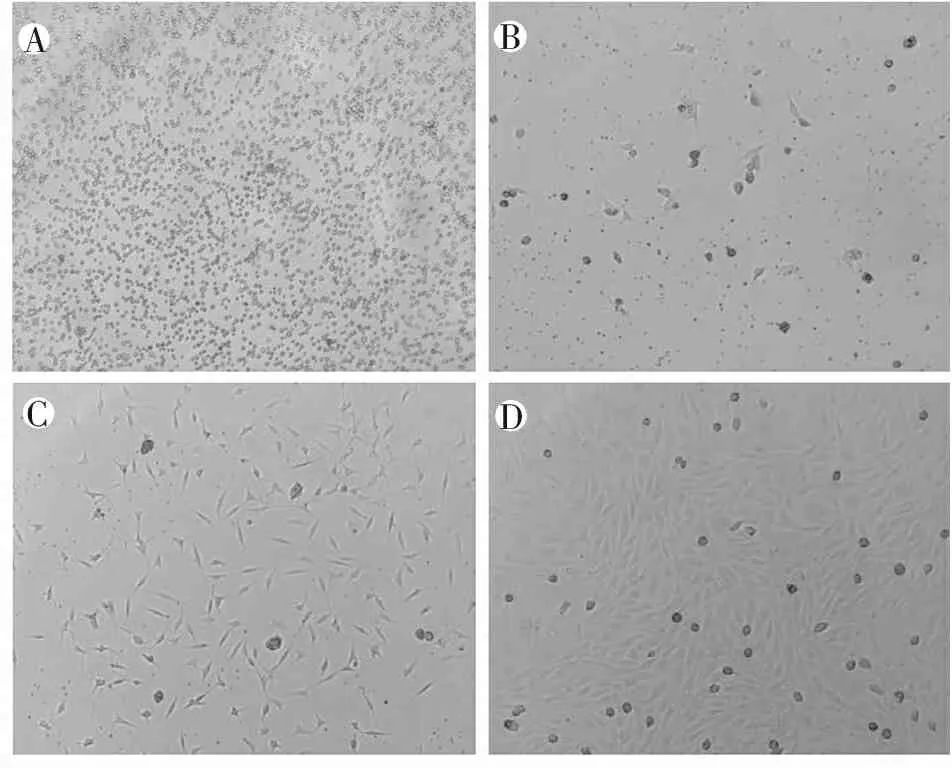
用EGM-2 BulletKit培养基定向诱导大鼠股骨和胫骨骨髓来源的单个核细胞14 d,最初细胞为悬浮状态,第3天可见少量梭形贴壁细胞,随后逐渐出现细胞集落,第7天细胞呈梭形或者多边形,第14天细胞融合度达到90%左右,呈铺路石样外观(图1)。
免疫荧光检测内皮细胞的表面标志物CD34对EPC进行鉴定,EGM-2 BulletKit培养基定向诱导培养14d后细胞CD34表达阳性率大于90%(图2)。上述结果表明,大鼠骨髓来源单个核细胞在用EGM-2 BulletKit培养基培养14d后,表现出内皮细胞特征。
2.2 APN慢病毒真核表达载体构建
以由大鼠EPC的总RNA逆转录出的cDNA为模板,PCR扩增得到APN基因的编码序列(图3a)。纯化的PCR产物(758 bp)被连接到慢病毒真核表达空载体pLKIRG上。连接后的表达载体(9 635 bp)用NheⅠ、 XmaⅠ双酶切检测(切割成745 bp、8 890 bp的两个片段),结果完全符合预期(图3b),并且比对测序结果,在 NCBI 中找到了与 APN基因编码序列完全一致的区域。
CBAD

CBA
2.3 APN过量表达细胞株构建
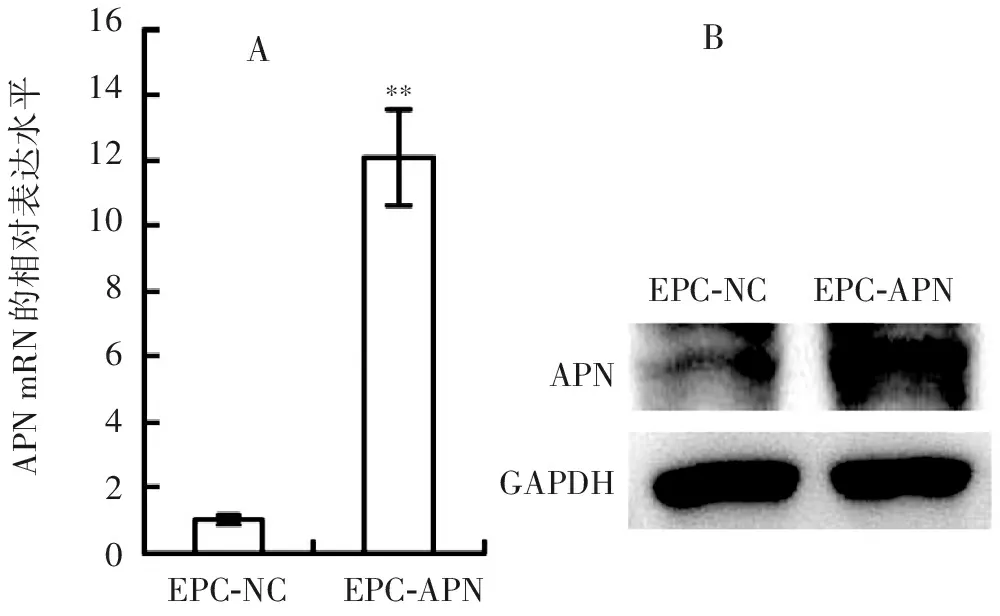
用APN慢病毒真核表达载体、空载体分别包装的慢病毒感染EPC,嘌呤霉素药筛3 d后收集细胞检测APN mRNA和蛋白的表达水平。检测结果显示EPC-APN中APN mRNA的表达水平显著升高,与对照组相比升高了约11倍(图4a),APN蛋白的表达水平明显增高(图4b)。
2.4 APN基因修饰对EPC增殖的影响
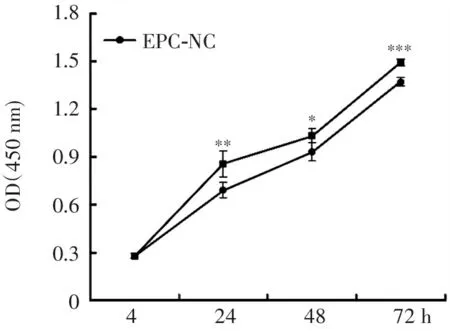
CCK8结果显示,在4 h时EPC-APN与EPC-NC的吸光度没有显著差异,在24、48 h和72 h时EPC-APN的吸光度显著增加,分别比EPC-NC的增加23.8%、10.9%和8.8%(图5),这表明在24、48 h和72 h时EPC-APN的增殖能力比EPC-NC的显著增加。
CBAD
1614121086420APNmRN的相对表达水平**EPC-NCEPC-NCEPC-APNEPC-APNAPNGAPDHAB
EPC-NC1.81.51.20.90.60.30OD(450nm)******2448724h
2.5 APN基因修饰对EPC迁移的影响
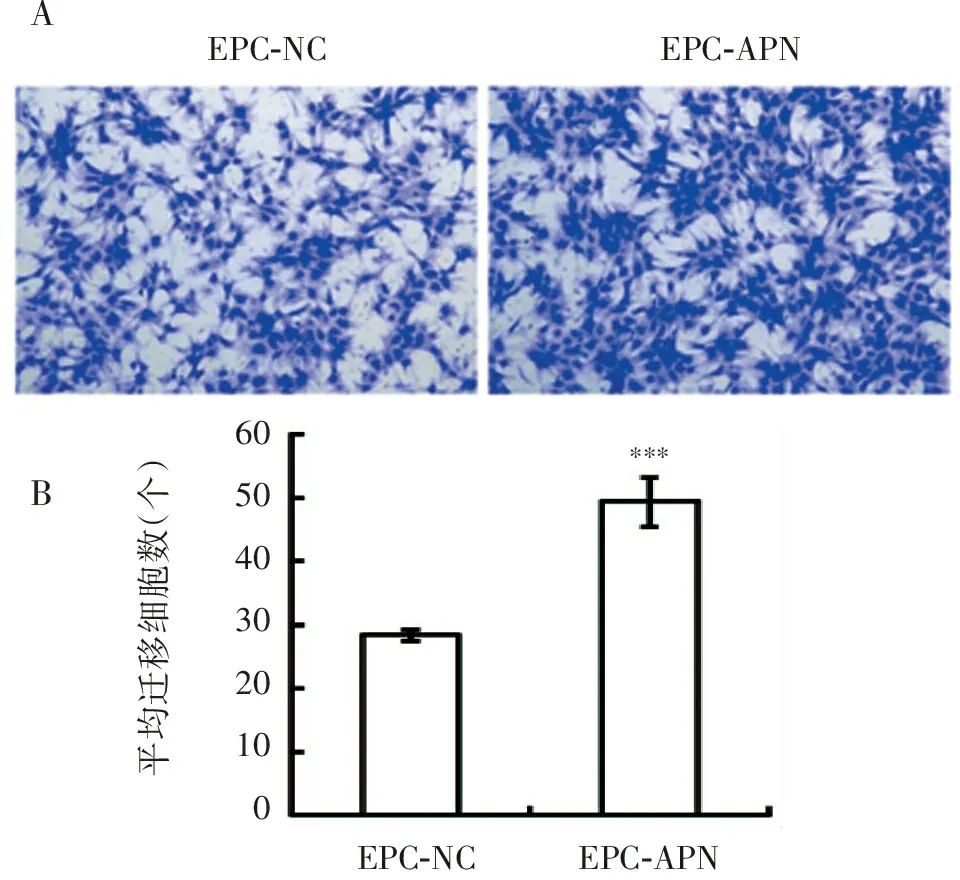
Transwell结果显示,与EPC-NC相比,EPC-APN的迁移能力显著增强,迁移到微孔膜下侧的EPC-APN的数量接近EPC-NC的2倍(图6)。
EPC-NCEPC-APN***EPC-APNEPC-NC6050403020100平均迁移细胞数(个)AB
2.6 APN基因修饰对EPC中VEGF、磷酸化VEGFR2、eNOS和磷酸化eNOS水平的影响
qPCR的结果表明,EPC-APN中VEGF、eNOS mRNA的表达水平较EPC-NC中的显著升高(图7a)。WB的结果表明,与EPC-NC相比,EPC-APN的VEGF、磷酸化血管内皮生长因子受体2(Vascular Endothelial Growth Factor Receptor 2,VEGFR2)、eNOS和Ser1177位点磷酸化eNOS的丰度明显增加(图7b)。
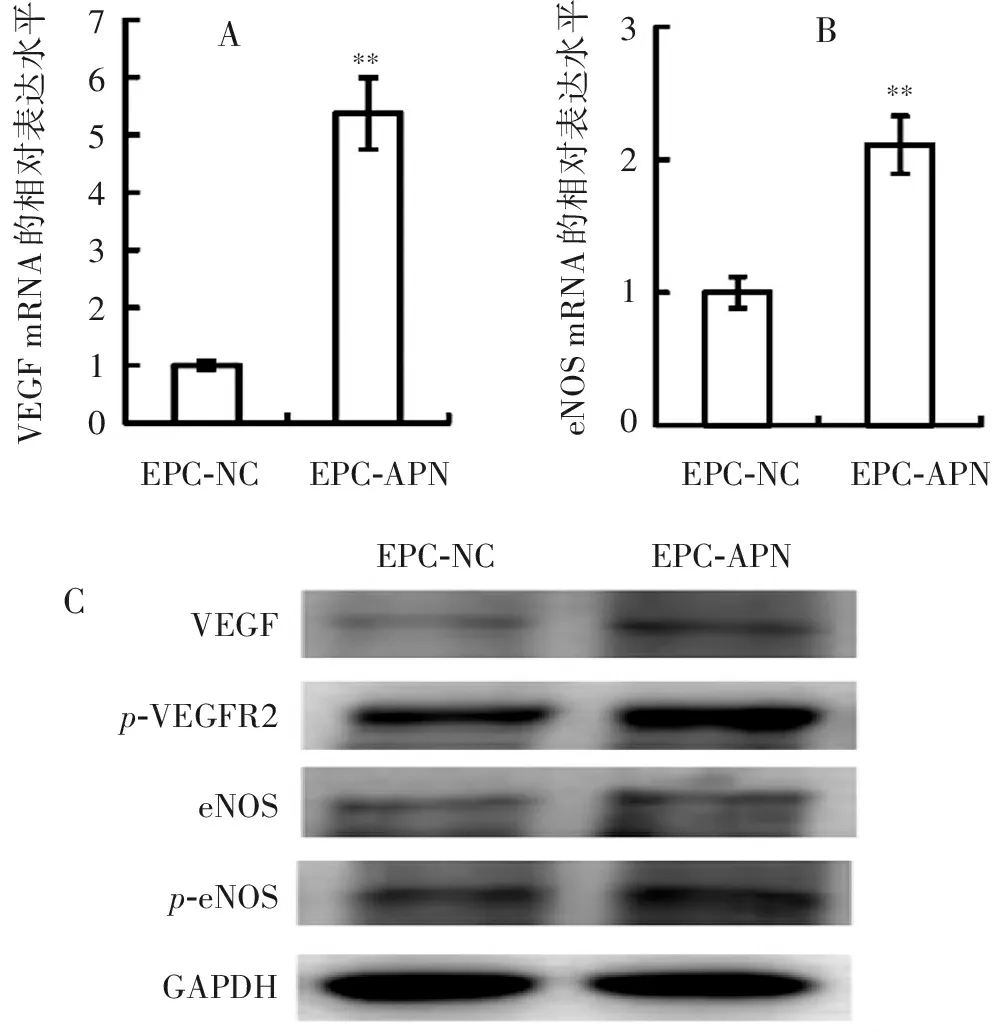
765432103210VEGFmRNA的相对表达水平eNOSmRNA的相对表达水平EPC-APNEPC-NCEPC-APNEPC-NC****EPC-APNEPC-NCVEGFp-VEGFR2eNOSp-eNOSGAPDHABC
3 讨论
本研究将大鼠骨髓来源的单个核细胞诱导成EPC,构建了APN基因慢病毒真核表达载体,通过慢病毒感染技术构建了APN基因修饰的EPC,并且研究了APN基因修饰对EPC增殖和迁移能力的影响及其机制。本研究的结果表明大鼠APN基因修饰增强大鼠骨髓来源的EPC的增殖和迁移能力(图5,6),这与在培养基中添加人源APN对人源EPC的影响一致[17,18]。APN基因修饰EPC的增殖和迁移能力更强,故对脑缺血的治疗效果比EPC更好[13]。图7表明APN基因修饰促进EPC中VEGF、eNOS表达,以及VEGFR2、eNOS Ser1177位点磷酸化。VEGFR2是内皮细胞中VEGF诱导信号转导的主要受体,结合配体时自磷酸化并被激活[23],磷酸化的VEGFR2募集互作蛋白,诱导下游信号转导[24]。APN基因修饰能够诱导EPC表达VEGF,故可以促进VEGFR2磷酸化。eNOS Ser1177位点磷酸化可以激活eNOS[22],因此APN基因修饰促进EPC中的eNOS活化。eNOS的表达水平和活性对EPC的迁移和血管生成至关重要[25,26]。由eNOS催化生成的NO通过亚硝基化和诱导VEGF表达促进EPC从骨髓中动员[25]。VEGF反馈促进eNOS在Ser1177处磷酸化,通过活化的eNOS动员骨髓来源的EPC[26,27],并且通过诱导Cx43表达促进EPC分化[21]。有研究表明APN通过AMPK信号通路诱导VEGF表达和eNOS Ser1177磷酸化[20,28],至于VEGF与eNOS哪一个在通路的上游,或者APN可以通过不同的信号通路诱导VEGF表达和eNOS Ser1177位点磷酸化尚需要进一步研究。
综上所述,本研究的结果表明APN基因修饰很可能通过促进EPC中VEGF、eNOS表达及eNOS活化增强EPC的活性。本研究为利用APN修饰的EPC治疗血管损伤性疾病和缺血性疾病提供了理论依据。
