基于DNA甲基化的分子亚型可预测肝细胞癌患者的预后
2020-12-21杨阮阮李锦忠龚晓兵
杨阮阮,李锦忠,龚晓兵
暨南大学附属第一医院感染科,广东 广州 510632
原发性肝癌是世界上第六大常见的癌症,也是导致癌症死亡的第三大原因,最常见的组织学是肝细胞癌(hepatocellular carcinoma,HCC)[1]。改善 HCC 患者预后一个重要阻碍是对HCC 的异质性不完全了解。目前的观点认为,每个原发性肿瘤的变异可能由遗传和表观遗传上不同的细胞组成,这导致每种肿瘤类型具有特定的表型异质性[2],在这些肿瘤细胞中有一些被称为肿瘤干细胞(cancer stem cells,CSC),其负责产生异质性肿瘤病变,也有可能助于治疗肿瘤的复发和转移。因此,通过对表观遗传的分子亚型进行临床特征及预后的分析对了解肿瘤异质性具有重要意义。
表观遗传是基因表达的可遗传状态而不改变DNA序列,甲基化是表观遗传学中最重要和最常见的修饰,也是调节基因组功能的重要手段[3]。全基因组甲基化水平降低是癌症早期的一种显著标志,并可能与癌症的严重程度及转移有明显关联。而特定基因启动子区域的CpG 岛异常高甲基化是癌症另一重要现象,导致染色体结构的改变并使肿瘤抑制基因和其他癌症相关基因沉默,使癌细胞能适应微环境并促进肿瘤发生和发展[4-5]。SHIN 等[6]分析了 HCC 和非肿瘤肝组织的甲基化谱,发现了HCC 中新的高甲基化基因,如TNFRSF10C 和IRF5 (参与细胞凋亡),HOXA9(胚胎发育过程中受调控)和NPY(参与细胞运动和细胞增殖)。此外,在肿瘤组织69 个显着低甲基化的基因中,已经鉴定出IL16 (趋化因子介导的免疫相关基因),BLK 和PGR 癌基因以及组蛋白去乙酰化酶1(HDAC1)。然而,尚未确定这些基因的启动子区域中的特定甲基化序列。因此,本文基于高通量组学数据整合了HCC的多个DNA甲基化生物标志物,开发了一种预后预测模型,以改善临床预后评估和个性化治疗。
1 资料与方法
1.1 数据获取和预处理 从TCGA 数据(https://cancergenome.nih.gov/)下载 level 3 级的 RNA-seq 数据(FPKM),包括374例肝细胞癌(Liver hepatocellular carcinoma,LIHC)患者样本和50 例癌旁样本。从UCSC(https://xenabrowser.net/)下载 430 例 LIHC 的 Illumina Infinium HumanMethylation450 阵列的甲基化数据。用 Perl(5.26)语言(http://www.perl.org/)的合并及 ID 转换脚本提取DNA甲基化矩阵和转录组矩阵,本研究仅包括来自临床随访时间超过30 d 的样本数据。每个位点的甲基化水平由β值表示,其范围为0(未甲基化)至1(完全甲基化)。使用sva R package[7]中的ComBat算法,通过整合所有DNA甲基化阵列数据,合并batch和patient信息来去除batch效应。
1.2 预后相关的甲基化位点筛选 首先,使用单变量Cox模型来计算每个异常甲基化基因的甲基化水平与患者总体存活(overall survival,OS)之间的关联,当P<0.05时,可以认为基因在单变量Cox分析中具有统计学意义[8]。然后,使用TMN 分期,病理分期(Stage),组织学分级(Grade),年龄和性别将从单变量COX比例风险回归模型中获得的显着CpGs引入多变量COX比例风险回归模型中,这些变量在单变量模型中也很重要。最后,选择在单变量和多变量Cox 回归分析中均显着的CpG位点作为特征性CpG位点。
1.3 甲基化肿瘤分型 使用R 中的Consensus Cluster Plus软件包[9]进行一致性聚类。根据最多可变的CpG位点识别LIHC子组。对应于一致性聚类的热图由pheatmap R 包生成。使用颜色渐变表示从0(白色)到1(深蓝色)的共识值;排列矩阵,使得属于同一簇的项目彼此相邻。在这种布置中,对应于完美共识的矩阵将显示颜色编码的热图,其特征是沿着白色背景上对角线的蓝色块。
1.4 生存和临床特征分析 Kaplan-Meier 图用于说明DNA 甲基化谱图定义的LIHC 亚组的总体存活率。使用对数秩检验来评估集群之间差异的显着性。使用R中的生存软件包进行生存分析。使用R的ggplot2 包进行肿瘤分型与临床特征的相关性分析。在所有测试中,P<0.05被认为差异具有统计学意义。
1.5 功能富集分析和基因组注释 将R 中的clusterProfiler 包[10]与 KEGG 结合使用,对预后位点所在的基因组中的基因本体论,生物途径和调控基序进行基因富集分析。
1.6 预后预测模型的构建和评估 R 中生存包的coxph函数用于基于CpG位点的甲基化图谱和预后信息的组合来构建Cox比例风险模型。模型的风险评分=CpG1*风险系数CpG1+CpG2*风险系数CpG2+CpGN*风险系数CpGN(“*”代表乘法)。Suivival、ROC 曲线和Calibration plot (校准图)判断风险模型的临床应用价值。
2 结果
2.1 筛选与OS相关的潜在预后甲基化位点 经对患者数据进行预处理后,确定了16 381个甲基化位点。然后使用单变量Cox 回归分析,在16 381 个甲基化位点中,有7 917 个CpG 位点被确定为肝细胞癌(LIHC)患者OS 相关的潜在DNA 甲基化生物标记物(P<0.05)。多因素独立预后分析筛选出具有独立预后能力的2 248个CpG位点。
2.2 不同的DNA甲基化预后亚组和簇间预后分析 使用2 248个潜在的预后甲基化位点的一致性聚类来识别LIHC 的不同DNA 甲基化分子亚组以进行预后分析。根据以下标准确定簇的数量:簇内的一致性较高,变异系数相对较低,CDF 曲线下的面积没有明显增加(图1A、1B)。根据类别数计算了平均聚类一致性和聚类之间的变异系数。累积分布函数(CDF)曲线下的面积在7 个类别后开始趋于稳定(图1B),最后确定最佳簇数为7,并制作了相关树状图表示定义明确的7 块结构及其一致性(图1C),另外TMN 分期,Stage,Grade,年龄,性别和 DNA 甲基化亚组作为注释,利用heatmap 函数生成与上述k=7 树图相对应的heatmap 图谱(图 2)。Kaplan-Meier 生存分析显示 7 个组之间的预后差异有显著统计学意义(P<0.001),聚类5和2的预后最佳,而聚类7和6的预后最差(图3)。然后,分别根据TMN 分期,Stage,Grade 和年龄、性别分析了7 个群集的群集内比例(图4)。相关临床特征与不同组聚类间的关联趋势如下:聚类4、6 和7 具有较高的病理分期阶段(Stage);T分期较低的集群为1、2和5;N 等级较高的集群为4 和6;性别总体以男性为主。本研究发现,不同临床特征在7 个群集中的所占比例各不相同,而且与集群的预后特征也相匹配(如聚类5的T分期较低而预后较好)。

图1 DNA甲基化分类的选择标准和一致性矩阵
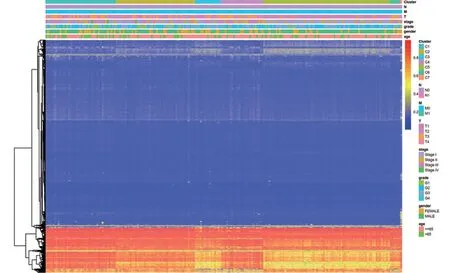
图2 以DNA 甲基化分级、TNM 分期、病理分期、组织学分级、性别和年龄为注释的heatmap图谱

图3 每个DNA甲基化亚型的生存曲线
2.3 基因功能富集分析和基因组注释 利用上述2 248个CpG位点的基因组注释,共鉴定出2 438个相应的启动子基因。然后对这2 438个基因进行功能富集分析,结果表明:在生物途径(biology process,BP),这些基因主要涉及ncRNA代谢过程、有丝分裂细胞周期相变的调控、细胞周期相变的调节、核糖核酸分解过程、信使核糖核酸分解过程。分子功能(molecular function,MF)主要参与催化活性,作用于RNA、核酸外切酶活性、单链DNA结合、核酸酶活性、催化活性,作用于tRNA。此外,细胞组成(cellular component,CC)主要涉及染色体区域、核斑点、染色体端粒的地区、浓缩的染色体和中心体(表1)。KEGG共发现24个相关的PATHWAYs(P<0.01),其中最集中且有意义的途径是RNA 运输、细胞周期、p53 信号通路和剪接体等(图5)。
2.4 构建和评估LIHC 预后预测模型 聚类5包含大量的样本和最多的特异性甲基化位点(图6),并且预后良好,因此选择它作为种子聚类。多变量Cox 回归用于构建肿瘤预后风险模型并发现8 个预后相关的甲基化位点:cg05489143、cg09600437、cg19165652、cg19569208、cg22732432、cg22958262、cg24153171、cg25455598 (表 2),由它们构建的风险模型可用于预后评估。获得的肿瘤预后风险模型是:风险评分=4.98*cg05489143-21.18*cg09600437+3.50*cg19165652+459*cg19569208+11.08*cg22732432+5.07*cg22958262-1.6.02*cg24153171 + 4.75*cg25455598。此外,根据上述模型计算的中位风险评分截止值,共有204 例患者被分为高风险组(n=101)和低风险组(n=102),LIHC的高低风险与甲基化程度的关系如图7,可以看到随着风险评分的增高,高风险组的生存时间虽然没有明显下降,但是死亡率明显升高。基于高、低风险分组采用Kaplan-Meier 方法绘制生存曲线(图8A),高低风险两组的预后差异具有显著统计学意义(P<0.001)。同时,ROC曲线的AUC值为0.822(图8B),提示该模型可以很好地预测患者的存活率。

图4 DNA甲基化簇之间的年龄、性别、TNM分期、病理学分期和组织学分级占比
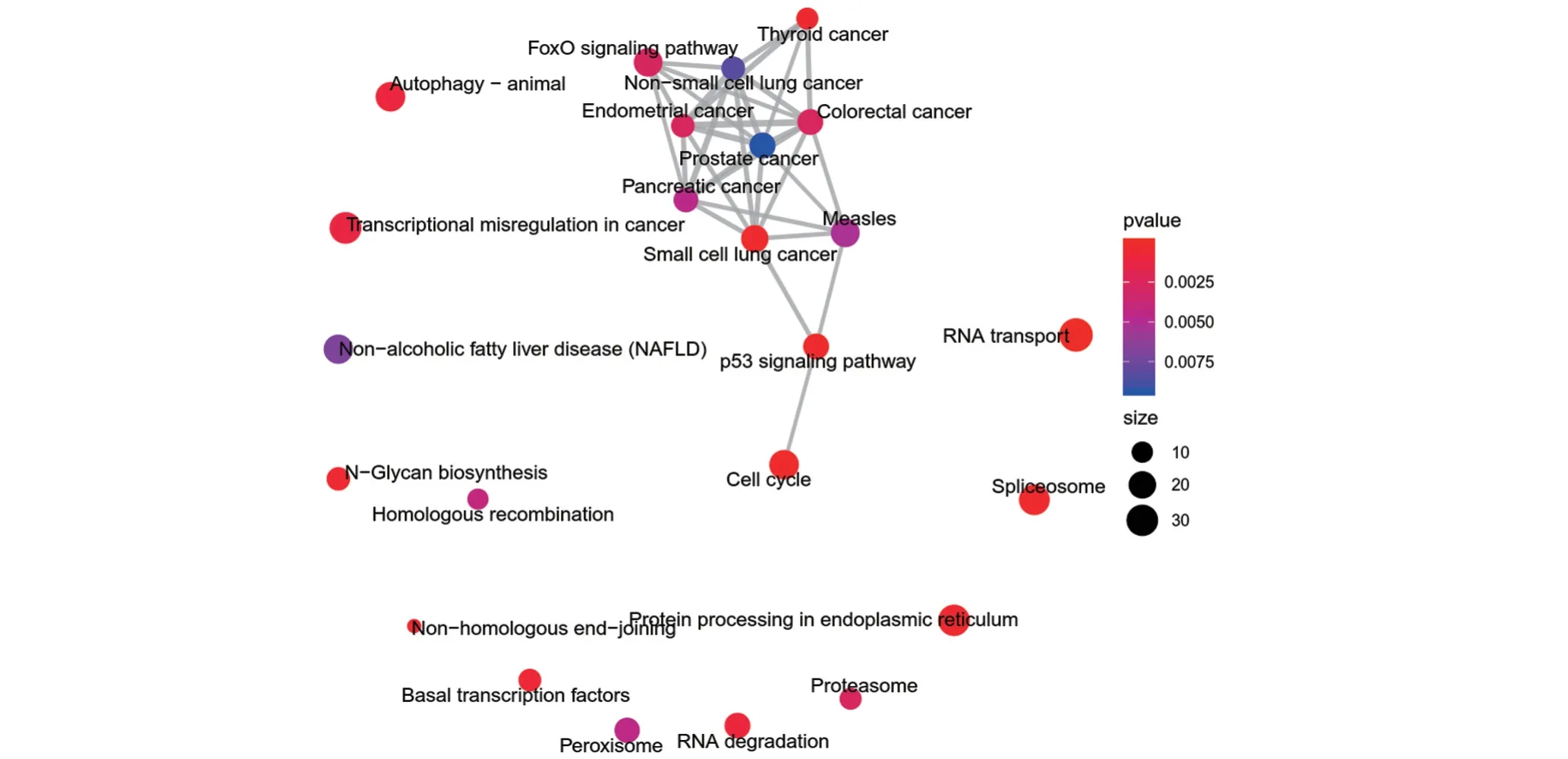
图5 对2 248个CpG位点的注释基因进行KEGG通路富集分析

表1 异常CpG位点的注释基因的GO功能富集
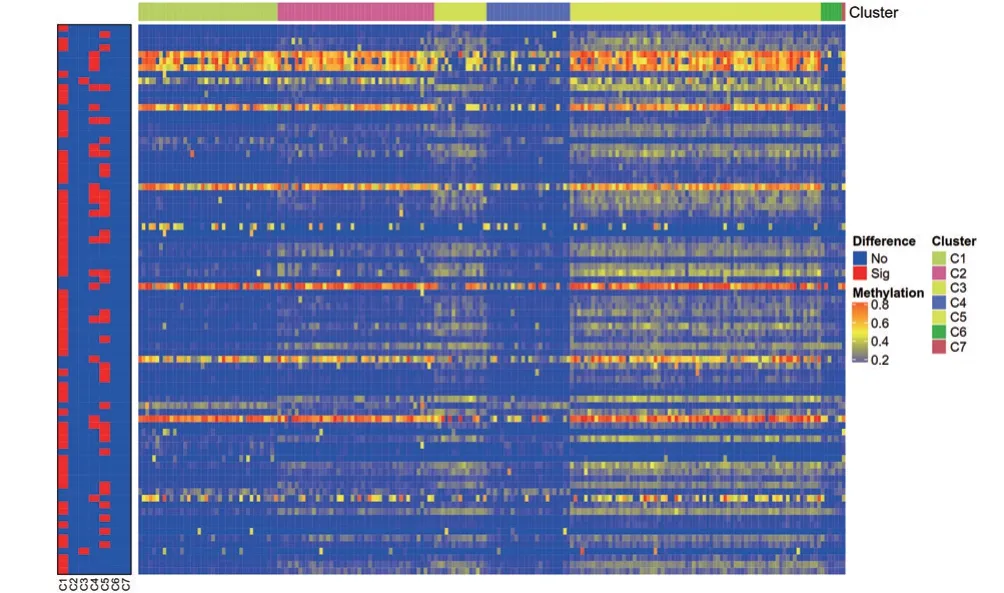
图6 每个DNA甲基化簇的特定的高/低甲基化CpG位点

表2 多变量COX回归得到的8个与OS显着相关的CpG位点

图7 高低风险组的热图分析

图8 模型对临床生存及预后的预测价值
3 讨论
近年来HCC的治疗取得了较大的突破,但肝癌的累积复发率分别在3年、5年时仍高达50%~60%、70%~100%[11]。尽管索拉非尼是目前HCC 最有效的靶向药物,也是唯一一种在晚期HCC使用的全身性靶向肿瘤治疗药物,但其疗效非常有限[12]。在这个前提下,HCC 中出现的表观遗传改变可能会成为一种新的治疗靶点[13]。DNA 甲基化作为表观遗传学中最重要和最常见的修饰,它是一种共价化学修饰,表现为胞嘧啶环的5位碳上加入甲基(CH3),这常见于5'CG3'这个基因序列中[14]。
本文研究了LIHC患者癌症样本和癌旁样本之间的异常甲基化位点,首先通过单因素和多因素COX比例风险回归分析最终筛选出具有独立预后能力的CpG 位点,接着利用一致性聚类方法进行肿瘤分型,最后根据分型差异分析构建甲基化位点的预后模型。根据该模型计算可以看到随着风险评分的增高,高风险组的生存时间虽然没有明显下降,但是死亡率明显升高。另外,高低风险两组的预后具有显著差异,同时,ROC 曲线的AUC 值为 0.822,表明模型可以很好地预测患者的存活率。有研究表明HCC 肿瘤会表现出与危险因素、肿瘤分期、分化程度和癌症治疗后存活相关的特异性DNA甲基化特征[15],并且大多数CpG 位点会倾向于从T1 期到T3 期逐渐高甲基化[16],本研究也发现临床分期等临床参数与集群的预后特征相匹配。近年来,已经有很多关于基因甲基化对各种癌症如肺癌[17],乳腺颈癌[18]和卵巢癌[19]诊断,治疗和预后评估的实验及临床研究。也有关于肝癌的相关研究,如CARM1介导的GAPDH甲基化是肝癌中葡萄糖代谢的关键调节机制[20]。另外,有实验证明P14ARF mRNA 水平受原发性肝癌中的DNA 甲基化调节,P14ARF基因DNA甲基化可能与HCC的发生及TNM分期有关[21]。与基因突变不同,表观遗传的改变具有可逆性,特别是DNA甲基化和组蛋白修饰[22]。通过使用去甲基化剂如5-氮杂胞苷(阿扎胞苷)和5-氮杂-2'-脱氧胞苷,可以实现DNA甲基化基因在癌细胞系中的重新表达,临床上在骨髓异常增生综合征和急性髓性白血病中的应用较常见和成熟[23-24]。因此,对基因甲基化的研究对未来肝癌的诊疗意义重大。
近年来,使用综合基因组工具的大规模基因组学和全基因组研究重塑了对癌症进化和异质性的理解。例如,RAN TAO 等[25]通过 HBV 相关 HCC 的全基因组甲基化谱发现7 个新基因(WNK2、EMILIN2、TLX3、TM6SF1、TRIM58、HIST1H4F 和 GRASP)在HCC 中高甲基化,在成对的相邻肝组织中低甲基化。SHEN等[26]也发现在HCC组织中有684个CpG位点显着高甲基化,这些基因中的5 个(CDKL2、CDKN2A、HIST1H3G、STEAP4、ZNF154)在高达63%的患者血浆中具有可检测的高甲基化DNA。鉴定的甲基化基因组可以是用于早期诊断的潜在生物标志物。但是以上这些研究只是从单个基因或多个基因的DNA 甲基化程度监测去评估在肝癌中的价值,肝癌的发病机制复杂,所以联合基因表达和DNA甲基化位点进行综合分析,并通过甲基化位点构建出了肝细胞癌的预后风险模型,把评估进行了量化分析,通过这个模型可以比较准确肝癌患者的生存情况,临床实用性更强。
综上,本研究在癌组织和癌旁组织中成功筛选出2 248个差异甲基化位点,DNA甲基化水平的差异与T分期、N 分期、M 分期、年龄、Stage、Grade 和预后的差异相关。接着利用一致性聚类方法得到7 个肿瘤亚组,亚组之间的预后有显著差异,同时对差异甲基化位点相应的启动子基因进行功能富集分析,它们主要涉及RNA运输、细胞周期、p53信号通路和剪接体,为肝癌发生机制提供了理论基础。最后根据分型差异分析构建甲基化位点的预后模型,该模型可以很好地预测患者的存活率。
