恩替卡韦片在中国健康人体中的生物等效性及安全性评价
2020-12-21柳正植高振月霍丹丹杨海淼
柳正植, 高振月, 任 庆, 霍丹丹, 杨海淼
1 长春中医药大学附属医院 Ⅰ期临床试验研究室, 长春 130021;2 正大天晴药业集团股份有限公司 临床中心, 南京 211122
全球有20多亿HBV感染者[1],流行率为2%~10%,是慢性肝炎、肝硬化和肝细胞癌的主要病因[2-3]。恩替卡韦能够抑制HBV所有的3个复制步骤,具有有效抑制病毒和抗药基因屏障的优势,已成为大多数HBV治疗指南推荐的一线选择[4-6],并已成为HBV治疗方案不可缺少的一部分[7-8]。近年来临床应用日趋广泛,且患者用药依从性较好[9-14]。恩替卡韦口服给药后1.5 h内可达血浆浓度峰值,Cmax和AUC与剂量呈线性比例增加。在稳定状态下,0.5 mg和1 mg片剂的Cmax均值分别为4.2~6.7 ng/ml和8.2~10.8 ng/ml。恩替卡韦主要通过肾脏消除,稳定状态下,清除率为给药剂量的62%~73%[7,15-16]。近年来,虽然恩替卡韦的药代动力学特性已被描述[16],但尚无在中国人群中研究新开发仿制药的生物等效性数据及安全性数据的报道。本研究旨在比较研究恩替卡韦仿制药与原研药恩替卡韦片在中国健康受试者中的生物等效性及安全性,为临床合理应用恩替卡韦片提供参考依据。
1 资料与方法
1.1 试验设计 本研究是一项随机、开放、两周期、两交叉、空腹状态下生物等效性试验。本次临床试验方案设计主要参考国家药品监督管理局2016年颁布的《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》[17]以及美国食品药品监督管理局2013年颁布的生物等效性研究药物的药代动力学行业指南[18]。本研究于2018年5月计划从长春中医药大学附属医院纳入28例健康受试者,受试者按照1∶1比例随机分配到2个给药序列之一。A组:第1周期受试者空腹服用受试制剂马来酸恩替卡韦片0.5 mg,经14 d洗脱期后,第2周期空腹服用参比制剂恩替卡韦片0.5 mg(Baraclude®);B组:给药流程与A组相反。
1.2 研究对象 年龄18~60周岁,男性或女性;男性受试者体质量≥50 kg、女性受试者体质量≥45 kg,体重指数18~28 kg/m2。志愿者生命体征、体格检查、实验室检查(血常规、肝功能、肾功能、乙型肝炎和丙型肝炎及艾滋病、梅毒筛查和尿常规等血液学、生物化学、尿检和血清学)、心电图及影像学等无显著异常。排除标准:药物过敏史;心血管、肝胆、肾脏、内分泌、血液学或胃肠疾病的病史或证据;试验前28 d内使用过任何肝药酶抑制剂或诱导剂,在研究前2周内使用任何处方药或草药;在研究前2个月内使用其他研究药物;在研究前48 h内摄入了咖啡因或巧克力的受试者;参与其他临床试验的受试者。
1.3 给药方案 经筛选合格的28例健康受试者服用受试制剂或参比制剂,通过SAS软件(9.2版本)随机分配受试制剂或参比制剂(依照给药随机表给药)。受试者于给药日早晨,空腹状态下口服受试制剂或参比制剂0.5 mg,用240 ml温开水送服。要求受试者在给药前1 d统一食用标准晚餐,在服药前至少禁食10 h,服药4 h后进食午餐,服药10 h后进食晚餐。
受试者住院观察至给药后第4天。根据恩替卡韦消除的平均终末半衰期(t1/2)约为128~149 h,药物累积指数约为每天1次给药剂量的2倍,表明其有效累积半衰期约为24 h。因此试验周期之间清洗期(给药间隔)设定为14 d,>10倍半衰期,理论上保证所有受试者下周期开始给药时,药物浓度低于生物分析定量下限,避免上个周期的处理影响到随后一周期的处理。
1.4 药代动力学评估 空腹状态下试验,每周期在给药前0 h(60 min内)及给药后10 min、20 min、30 min、45 min、1.0 h、1.5 h、2.0 h、3.0 h、5.0 h、8.0 h、12 h、24 h、36 h、48 h、72 h共16个时间点进行药代动力学分析。全血样本在2~8 ℃、1700 g离心力条件下离心10 min,获得血浆,(-70±10)℃条件下保存,以供药代动力学分析。
1.5 分析方法 采用经验证的、特异的、灵敏的液相色谱-串联质谱法(LC-MS/MS)测定canagliflozin的血浆浓度[19]。蛋白用甲醇溶液沉淀后,进行色谱分析。色谱柱为Atlantis T3 C18 3 μm,50×2.1 mm。流动相以流动相A:5 mM乙酸铵(NH4AC)水溶液;流动相B:甲醇(MeOH);流速:0.600 ml/min。进样体积:5.00 μl;柱温:40 ℃。质谱分析采用Triple Quad 6500(Applied Biosystems/MDS Sciex)。有效定量线性范围为0.0400 ng/ml至10.0 ng/ml。
1.6 安全性评价 安全性评估包括基于给药后的临床和实验室检查不良事件(包括受试者报告的所有主观症状和研究人员观察到的客观体征)。按照药物种类列表描述不良事件,统计不良事件发生率。
1.7 伦理学审查 本研究方案经由长春中医药大学附属医院伦理委员会批准,批号:CCZYFYLL2018-审字006,受试者在参与本研究前均签署书面知情同意书。
1.8 统计学方法 药代动力学参数采用WinNonlin 7.0版本的非房室模型,按照预定的时间点采集测定的血样数据,当实际采样时间超出采血窗,采用实际采血时间计算主要药代动力学参数。采用线性和半对数做图来绘制完成试验的受试者空腹口服受试制剂及参比制剂后的恩替卡韦的平均血药液度-时间曲线,每一名受试者的血药液度-时间曲线也一一报告,通过描述性统计对各药代动力学参数进行分析。按照给药分组,对主要药代动力学参数计算、均数、标准差、中位数、最小值、最大值、几何均数、变异系数(CV)。对药代动力学参数AUC、Cmax和Tmax进行统计分析,显著性水平为5%。AUC和Cmax需进行对数转换后进行方差分析。方差分析包括以下因素:制剂因素、周期和序列。将主要药代动力学参数Cmax、AUC0-t、AUC0-∞经过对数转换后,进行方差分析、双单侧t检验和90%CI分析,Tmax经非参数检验,评价受试制剂和参比制剂Tmax的差异。当受试制剂与参比制剂的Cmax、AUC0-t、AUC0-∞几何均数比的90%CI在80.00%~125.00%等效区间内,认为两制剂生物等效。
2 结果
2.1 受试者入组及完成情况 纳入78例受试者,按照纳入排除标准筛选出28例合格受试者,均符合试验方案要求,进入全分析集(表1,图1)。
2.2 药代动力学和生物等效性
2.2.1 药-时曲线 28例健康受试者口服0.5 mg恩替卡韦片的受试制剂与参比制剂后,所得不同时间的平均血药浓度-时间曲线,结果显示,受试制剂和参比制剂达峰时间和血药峰浓度基本一致(图2~3)。

表1 28例受试者一般资料

图1受试者分布流程图
2.2.2 药代动力学参数比较 受试制剂与参比制剂的体内过程一致。空腹状态下等效性判定结果显示符合生物等效性集的28例受试者单次服用恩替卡韦受试制剂或参比制剂后,Cmax、AUC0-t、AUC0-∞几何均数的比值分别为98.18%、101.97%、103.07%,其90%CI分别为91.36%~105.50%、98.32%~105.74%、96.30%~110.32%,均落在80.00%~125.00%之间。对药代动力学参数Cmax、AUC0-t、AUC0-∞经自然对数转换后,方差分析结果显示,空腹状态下给药周期间差异有统计学意义(P<0.05),给药序列及制剂因素差异均无统计学意义(P>0.05)。符合生物等效性判定标准(表2~4)。
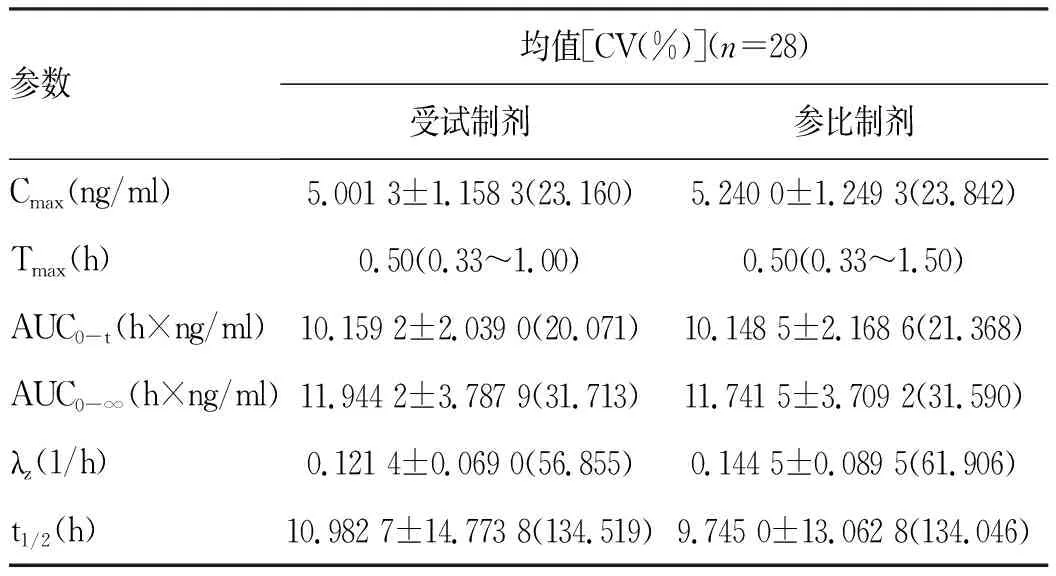
表2 空腹状态下受试制剂和参比制剂主要药代动力学参数
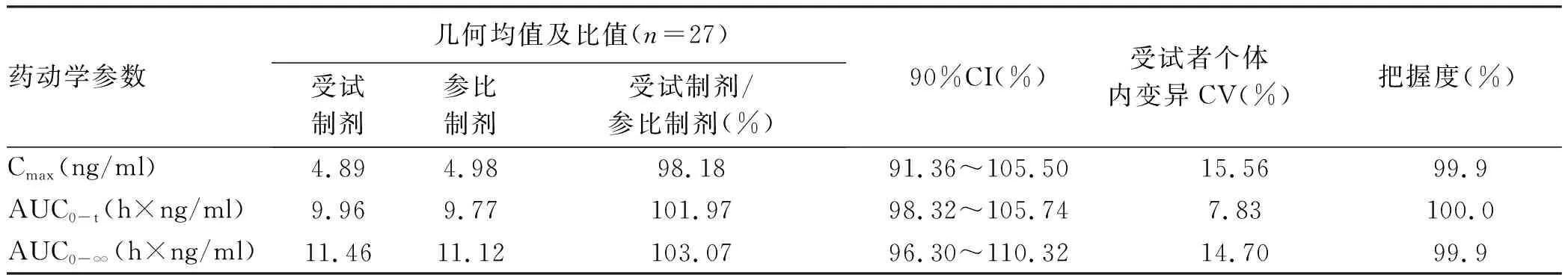
表3 受试制剂和参比制剂等效性判断结果
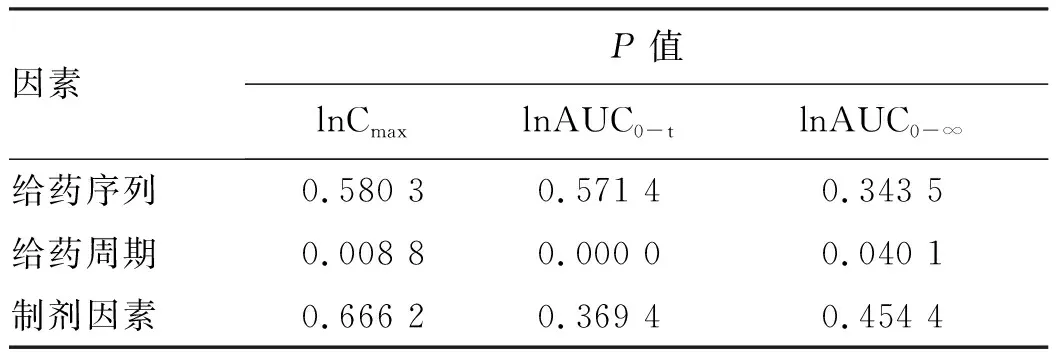
表4 固定因素方差分析结果
2.3 安全性评价 全部进入安全性数据分析集的28例受试者中,共5例受试者发生6例次不良事件,未发生严重不良事件及非预计严重不良事件,未发生死亡。从安全性角度看,受试制剂恩替卡韦片与参比制剂恩替卡韦片(Baraclude®)安全性均较好,且受试制剂的不良事件发生率相对更低(表5~6)。
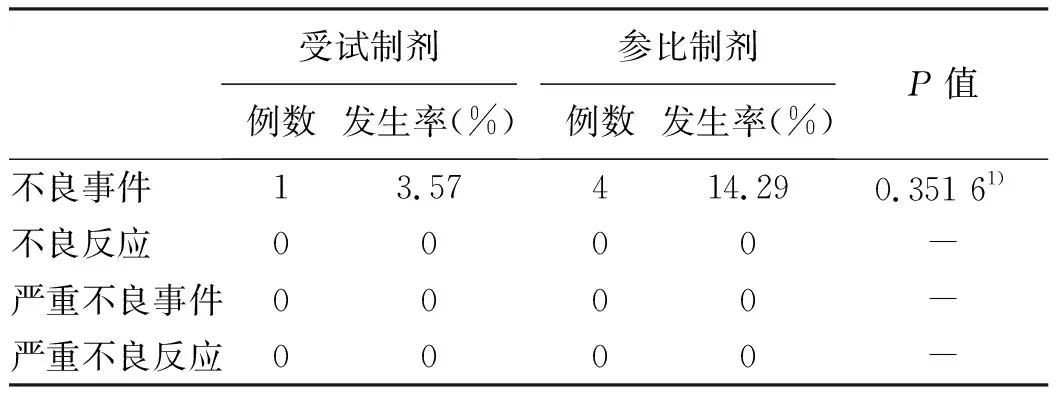
表5 受试制剂和参比制剂各类不良事件/不良反应发生情况
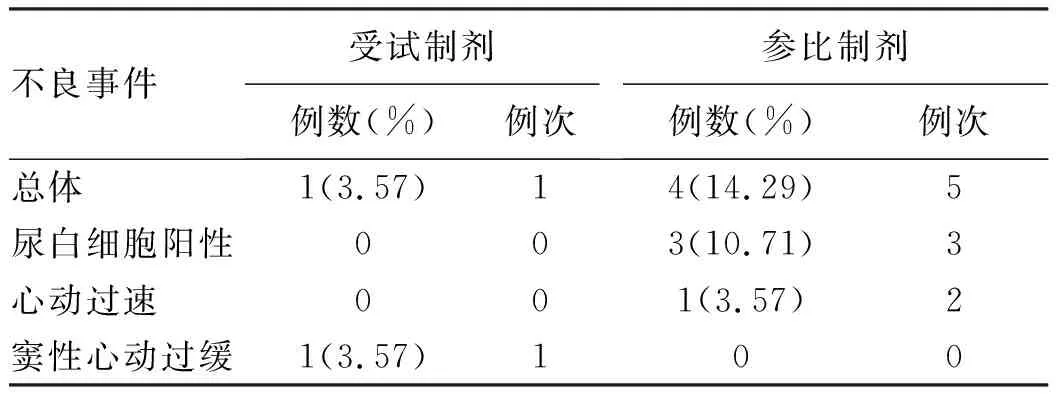
表6 不良事件发生情况
3 讨论
临床上,恩替卡韦主要用于病毒复制活跃、血清ALT水平持续升高或肝组织学显示有活动性病变的慢性乙型肝炎的治疗。本研究中,两种剂型在所有受试者中耐受良好,未发生可能影响研究结果的意外事件。口服0.5 mg恩替卡韦后,平均血浆浓度-时间曲线显示,受试制剂和参比制剂达峰时间与血药峰浓度基本一致。生物等效性研究最重要的目标是保证试验品和对照品的安全性和有效性,一般认为,Cmax、AUC0-t、AUC0-∞等基本药代动力学参数的等效范围为80%~125%。本研究结果显示,受试制剂和参比制剂的Cmax、AUC0-t、AUC0-∞无统计学差异,表明二者具有可比性。血浆中恩替卡韦的主要药动学参数Cmax、AUC0-t、AUC0-∞几何均数比值的90%可信区间均在80.00%~125.00%,符合指南[18]规定的生物等效性范围。从吸收的程度和速率来看,与参比制剂相比,受试制剂在中国受试者人群中是生物等效的。此外,受试制剂和参比制剂总体上安全性良好,但有3例受试者服用参比制剂后出现尿白细胞水平升高的不良事件,这种情况在已知的恩替卡韦不良反应和相关文献报道中均未见报道,值得关注。
恩替卡韦生物等效性的研究只局限于健康受试者单剂量给药,尚缺乏针对乙型肝炎患者的研究,国外已有恩替卡韦在乙型肝炎患者中的药代动力学研究[20]。国产仿制药在原辅材料、工艺水平、技术条件、质量管理等方面与进口药品存在一定差异,但这种差异对实际临床疗效的影响尚缺乏循证医学证据探讨,尚须对国产仿制药品的长期用药安全性、抗病毒效果、耐药发生率以及患者依从度等方面进一步归纳总结。
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突,特此声明。
作者贡献声明:柳正植、杨海淼、高振月负责课题设计,资料分析,撰写论文;任庆、霍丹丹参与收集数据,修改论文;柳正植负责拟定写作思路,指导撰写文章并最后定稿。
