GC-MS/MS分析中药六方藤和山苦荬中46种禁限用农药残留量
2020-12-16唐明华蒋永宁卢桂健
唐明华,蒋永宁,徐 梦,卢桂健,梁 爽
(1.玉林市食品药品检验检测中心,广西 玉林 537000;2.广西中医药大学药学院,南宁 530001)
六方藤(Cissus hexangularisThorel ex Planch)为葡萄科白粉藤属植物翅茎白粉藤的藤茎[1],具有祛风除湿,活血通络之功效,临床用于风湿关节痛,腰肌劳损,跌打损伤[2]。现代药理学研究表明,白粉藤属植物具有多种潜在的抗菌、抗炎、抗氧化、抗过敏、抗肿瘤、拮抗内皮素和治疗糖尿病等生物活性[3]。六方藤化学成分复杂,含有糖类、苷类、皂苷类、鞣质、黄酮类、酚类、蒽醌类、香豆素、强心苷、甾萜类、生物碱及挥发油等化学成分[4]。山苦荬[Ixeridium chinense(Thunb.)Tzvel.]为菊科小苦荬属多年生草本植物[1],以全草入药,具有清热解毒、止痛消肿、消炎凉血等功效,用于治疗无名肿痛、腹腔脓肿、痢疾、关节炎等多种疾病[5]。现代药理研究证明,山苦荬的化学成分具有抗炎保肝、抗氧化、抗烟碱、抗病毒、抗白血病等作用,其中黄酮类成分具有很强的药理活性[6]。近10年来,国内外学者从山苦荬中分离鉴定出了倍半萜类、三萜类、甾醇类、黄酮类等生物活性化学成分[7]。在中药六方藤和山苦荬生长过程中,为消除病虫草害,提高产量和品质,喷洒农药必不可少,然而施药过程的不规范和不合理,常常使药材中农药残留超标[8]。为保证消费安全,国家加大了对药材中农药残留的监测力度,2020版《中国药典》四部通则将33种禁用农药正式列入《0212药材和饮片检定通则》,不断增多的检测项目和日益严格的限量要求使农药残留检测工作面临更加严峻的考验[9]。由于气质联用仪具有高分辨率和高灵敏度,因此被广泛应用于分离与鉴定复杂组分,是中药中农药残留定性定量的最有效工具之一[10,11]。试验在中药日常检验项目的基础上,剔除在质谱上灵敏度较差的项目,最终确定46种农药品种作为目标物。通过对前处理方法的提取溶剂、洗脱溶剂等方面进行优化,建立了准确、可靠且能同时测定中药六方藤和山苦荬中46种常见禁限用农药的多残留检测方法。
1 材料与方法
1.1 材料与仪器
1.1.1 仪器 TRACE 1310-TSQ8000 Evo气相色谱质谱联用仪,配有电子轰击源(EI),美国Thermo Fisher公司;BSA822-CM型电子天平,赛多利斯科学仪器(北京)有限公司;ROATNTA 460R型离心机,德国SIGMA公司;HYQ-3110型涡旋混匀器,美国精骐公司;N-EVAP-24型氮吹仪,美国Organomation公司。
1.1.2 材料和试剂 农药标准品:环氧七氯,德国Dr.公司,批号G121441;46种农药混标溶液,100.0 μg/mL,First Standard公司生产,批号S001417,包括α-六六六、β-六六六、γ-六六六、δ-六六六、苯醚甲环唑、丙溴磷、哒螨灵、对硫磷、敌敌畏、虫螨磷、对硫磷、二甲戊灵、二嗪磷、伏杀硫磷、氟胺氯菊酯、氟氯氰菊酯、腐霉利、甲胺磷、甲拌磷、甲拌磷砜、甲基对硫磷、甲基异柳磷、甲氰菊酯、久效磷、乐果、联苯菊酯、磷胺、硫丹、高效氯氟氰菊酯、氯氰菊酯、马拉硫磷、咪酰胺、醚菌酯、氰戊菊酯、三氯杀螨醇、三唑磷、三唑酮、杀螟硫磷、水胺硫磷、五氯硝基苯、溴氰菊酯、亚胺硫磷、氧乐果、乙烯菌核利、乙酰甲胺磷、异菌脲。乙腈、正己烷,色谱纯,美国Thermo Fisher公司;无水硫酸钠,分析纯,广州化学试剂厂,用前在650℃灼烧4 h,贮于干燥器中,冷却后备用;QuECh-ERS萃取盐包,美国Agilent公司,部件号5982-6755(含6 g无水MgSO4,1.5 g NaAc);15 mL QuECh-ERS分散固相萃取管,美国Agilent公司,部件号5982-5456(含300 mg PSA,300 mg C18,90 mg GCB,900 mg无水MgSO4)。20批次中药六方藤和山苦荬药材样品分别为卫矛科雷公藤属植物六方藤和菊科小苦荬属的山苦荬全草,样品信息见表1。
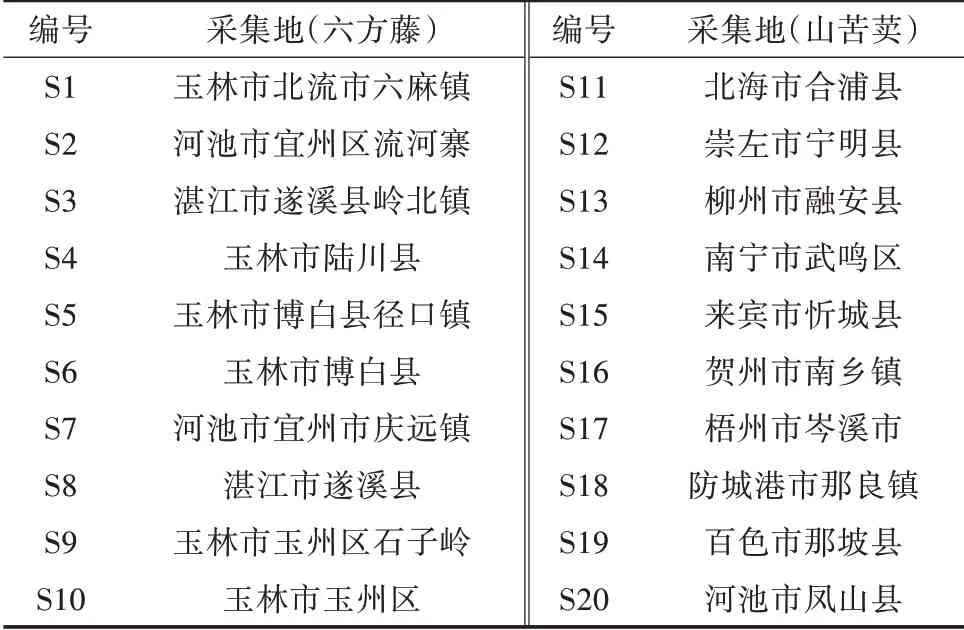
表1 六方藤和山苦荬样品基源信息
1.2 方法
1.2.1 标准溶液的配制 精密量取1.0 mL 46种农药混标溶液,置于20 mL容量瓶中,用丙酮溶解并稀释至刻度,摇匀,即得混合标准储备液(浓度均为5.0μg/mL)。再精密量取2.0 mL置10 mL容量瓶中,用丙酮定容,即得混合标准中间液(浓度为1.0 μg/mL)。分别精密量取0.10、0.20、0.30、0.40、0.50、0.60 mL混合标准中间液于6个2 mL容量瓶中,加乙腈定容至刻度,再加入0.1 mL浓度为1.0μg/mL的内标容液,摇匀,即得系列标准工作溶液(浓度依次为50.0、100.0、150.0、200.0、250.0、300.0 ng/mL,内标浓度为50.0 ng/mL)。
1.2.2 供试品溶液的制备
1)提取。称取样品2 g,于50 mL的聚苯乙烯具塞离心管中,加入1%冰醋酸溶液15 mL,涡旋使药粉充分浸润,放置30 min,精密加入乙腈15 mL涡旋振荡5 min,加入QuEChERS萃取盐包,立即摇散,涡旋振荡3 min,置冰浴中冷却10 min,5 000 r/min离心5 min。
2)净化。取上清液9 mL置于15 mL QuECh-ERS分散固相萃取管中。涡旋使充分混匀,再置振荡器上剧烈振荡5 min使完全净化,离心5 min,精密吸取上清液5 mL,置氮吹仪上40℃水浴浓缩至近干,加入内标溶液0.1 mL,加乙腈定容至2 mL,涡旋混匀,经微孔滤膜(0.22μm)过滤,取续滤液,装瓶,即得。
1.2.3 气相色谱-串联质谱分析条件
1)色谱条件。TG-5MS石英毛细管柱(30 m×250μm×0.25μm)。载气为He(≥99.99%),柱流量1.2 mL/min,进样口温度270℃,不分流,进样量1μL,溶剂延迟3.0 min。程序升温:初始温度40℃,保持1.5 min,以25℃/min升至90℃,保持1.5 min,以25℃/min升至180℃,以5℃/min升至280℃,以10℃/min升至300℃,保持5 min。
2)质谱条件。EI源,电子能量为70 eV,灯丝电流25μA,离子源温度为300℃,质谱接口温度为280℃,多反应监测(SRM)采集模式。Trace Finder软件自带的数据库包含超过1 000种能在GC上分析的常规化合物,每种化合物都有3对离子对,其中响应最高的一对为定量离子,其他两对为定性离子。每个化合物SRM质谱条件(母离子-子离子-碰撞能量)可以从Trace Finder软件中的数据库直接导出编辑进样方法和数据处理方法。46种农药的保留时间、SRM离子对信息、碰撞能见表2,目标物的SRM总离子流见图1。

表2 46种农药的保留时间、SRM离子对信息和碰撞能

图1 混合标准工作溶液的总离子流
2 结果与分析
2.1 方法学验证
2.1.1 标准曲线、检测下限和精密度 将46种农药标准溶液按试验条件进行测定,以农药的质量浓度为横坐标,峰面积为纵坐标进行线性回归,以信噪比(S/N)=3确定检测下限。结果表明,46种农药在50.0~300.0 ng/mL与目标物的响应值呈良好的线性关系,相关系数(r2)均大于0.990,方法检出限为0.01~0.60μg/kg。取溶液连续进样6次,以各指标色谱峰积分面积RSD计算精密度,精密度良好(RSD<10%)。各指标线性方程、精密度、相关系数、检出限结果见表3。

表3 46种农药的线性方程、相关系数及定量下限
2.1.2 加标回收率 加标回收率试验设置高、中、低3个浓度水平,各浓度水平分别准确称取空白样品5.0 g,分别精密加入“1.2.1”项下的混合标准品储备液0.06、0.12、0.24 mL,按照“1.2.2”制备加标回收样品溶液,每个级别平行制备2份,分别进样测定,计算加标回收率和相对标准偏差(RAD),结果见表4。46种农药的平均回收率在48.6%~119.0%,相应的RAD为0.1%~14.9%。
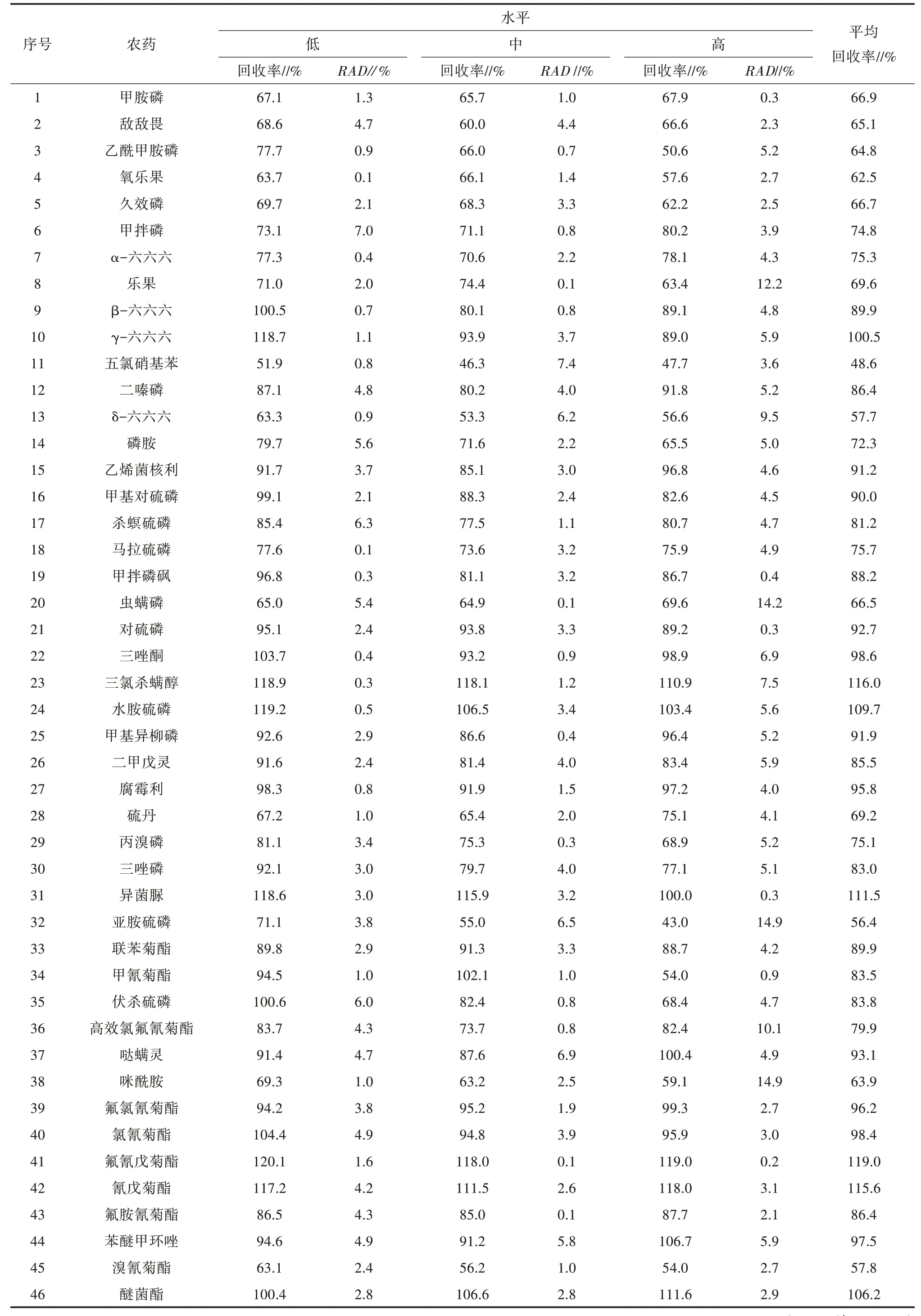
表4 加标回收结果
2.2 样品测定结果
试验采用上述样品前处理方法以及色谱分离条件、质谱检测条件检测了20批次六方藤和山苦荬样品。结果表明,46种禁限用农药检测指标在10批山苦荬样品中最终检出5种,分别为虫螨磷、高效氯氟氰菊酯、氯氰菊酯、二甲戊灵、苯醚甲环唑。虫螨磷在样品S13、S16、S17中均有检出,残留量有较大差别,除S16外,其他样品均低于5μg/kg,S13检测出高效氯氟氰菊酯8.25μg/kg,S18检测出二甲戊灵12.03μg/kg,S16分别检测出虫螨磷28.89μg/kg,氯氰菊酯19.52μg/kg,苯醚甲环唑24.30μg/kg。除S14外,其他样品均低于5μg/kg;六方藤样品中尚无检出。
3 讨论
中药山苦荬及六方藤中农药残留分析属于痕量分析,其技术特点主要有样品中农药的残留量低,而干扰物如色素、挥发油等含量高,给测定带来较大难度;农药品种繁多,用药情况复杂,各类农药性质差异较大。针对上述特点,分析时分4步进行,第1步,确定测定目标,调查山苦荬和六方藤农药污染情况,包括种植区环境特点、农药使用习惯和用药历史、轮作作物农药使用情况和常见病虫害及其药物防治方法。第2步,确定分析方法。色谱串联质谱分析技术是目前最重要、应用最广泛的多农药残留检测技术。以气相色谱进行分离,以灵敏度高、专属性强的串联质谱仪作为检测器测定,定性与定量可同时实现。建立分析方法时有3个方面需要优化:①优化仪器采集参数,对于GC-MS/MS方法,需要优化的参数主要包括毛细管色谱柱的选择、载气流速、柱温箱升温程序、离子源参数、离子对选择、碰撞电压优化和数据采集模式等;②优化样品前处理方法,根据样品基质的性质,尽可能减少干扰物质,保留目标化合物。其中提取溶剂选择(乙腈、丙酮)、提取方法(振荡法、超声波提取法)、净化方法(固相萃取柱净化法、固相分散萃取)是关键点,直接影响加标回收率及方法检出限和定量限;③采用适宜方法补偿基质效应。样品前处理方法会改善基质效应,为了抵消基质效应对定量准确性的影响,一般采用空白基质匹配法。第3步,方法学验证。主要包括线性、精密度、重复性、回收率、检测限与定量限等。第4步,测定样品。样品在采集、制备、检测时应始终保持其原始特性,未受污染、变质或混淆,尤其应当注意采样代表性和取样均匀性。检测所得结果应由保留时间和碎片丰度共同作为判定依据。
