结构决定性质:材料基因组学的“三观”
2020-12-15陈砚美田正芳甘喜武
徐 涛,陈砚美,田正芳,甘喜武,付 军
(1. 黄冈师范学院 化学化工学院,湖北 黄冈 438000;2. 黄冈市教育局,湖北 黄冈 438000;3. 湖北省黄冈中学,湖北 黄冈 438000)
化学研究的是构成宏观物体的物质。对物质的研究可分为物质的组成与结构和物质的性质与变化两个方面。物质的组成与结构决定了物质的性质与变化;物质性质的改变是物质的组成与结构发生了变化的结果。因此,分子结构决定材料性能,材料性能决定材料应用。
材料是人类文明大厦的基石。现代社会,经济社会的发展和科学技术的革新都越来越依赖于新材料的发现和应用[1]。2016年国务院印发的《“十三五”国家战略性新兴产业发展规划》和2018年工信部、财政部联合印发的《关于印发国家新材料产业资源共享平台建设方案的通知》中都明确指出,新材料是国民经济先导性产业和高端制造及国防工业等的关键保障,是我国战略竞争的关键布局。新材料,是相对于传统的材料而言,是指新近发展或正在发展的材料,主要体现在具有更加优异的性能和特殊的性质,如新型陶瓷材料、超导材料和隐形材料等等。传统材料的发现,往往是类似于爱迪生寻找合适灯丝材料那样,遍及所有已知材料进行一一尝试,这种方法不仅费时费力,同时也受限于已知材料的范围。为了探索未知的材料世界,类比于生物基因组学,材料基因组学这一概念应运而生[2]。
1 结构决定性质
分子是保持物质化学性质的最小微粒。分子是由原子构成,因此物质的元素组成决定了物质的性质,如铁易生锈,真金却不怕火炼。此外,分子的组成也决定了物质的性质,比如CO易燃,CO2却能灭火。晶体结构是决定物质性质的又一个重要因素,最简单的例子莫过于同为碳单质的石墨与金刚石,前者低廉柔软,后者却珍贵坚硬。了解人类探索物质结构的过程,认同“物质结构的探索是无止境的”观点,进而从原子、分子、超分子等不同尺度认识物质结构的意义。此外,在一定条件下,物质的聚集状态随构成物质的微粒种类、微粒间相互作用、微粒的聚集程度的不同而有所不同。物质的聚集状态会影响物质的性质,通过改变物质的聚集状态可能获得特殊的材料。
通过计算机辅助材料分子结构的研究是一种十分有效的选择,不仅能够处理已知材料,而且还能够通过修饰分子结构从而预测未知材料分子的性能。此外,由于处理过程都是通过计算机辅助完成,几乎零成本的资源消耗,避免了传统化学工作者费时费力的实验过程。为了整合国内外材料研究的成果,发现未知的新材料,很有必要建立、完善和补充材料数据库。
2 材料基因组学
材料基因组学是材料与信息科学的交叉学科[3]。为了改变传统材料寻找的局限性,在已知数据库和模型的基础上,借助计算机来加快材料创新和筛选过程,从而获取更加优质的材料。近年来,材料基因组学受到了学术界的重视[4-6],中科院物理所、南京大学物理学院,以及美国普林斯顿大学的团队的三个独立工作同时发表在2019年2月28日的《Nature》期刊上,尤其需要关注的是南京大学物理学院万贤纲教授团队发表的《利用对称性指标进行拓扑材料的广泛研究》的论文[6],《Nature》编辑部指出该项成果“使得新奇的拓扑现象离应用更近了一步,或可引发电子学和催化学等方向的革命。”与寻找传统材料相比,其优势主要体现在:(1)通过计算机进行模拟和计算,而无需耗费大量时间和资源来做实验;(2)由于是在计算机上进行操作,其成本接近于零,因此可以对材料进行修饰创新,或者对多个候选材料进行组合排列[7]。材料基因组学的本质是机器学习,其基本原理如图1所示。首先通过收集已知材料的特征、属性、自变量和预测变量等作为输入术语,再通过在机器上进行建模、算法和技术处理,从而获得种类、目标、因变量和响应变量等输出术语。材料基因组学致力于预测使用量子力学的材料特征,并在计算机上进行仿真。

图1 机器学习揭开有机电子基因组新材料应用的奥秘Fig.1 Machine learning reveals the mystery of the application of new organic electronic genome materials
2.1 微观之分子结构
微观粒子,一般指的是直径小于10-9米(即纳米以下)的微粒,常见的原子、分子都属于这一范畴。原子的基本性质(如原子半径、原子质量、核电荷数、核外电子排布、电离能和电负性等)和分子的结构与性质(如分子的立体结构和分子之间的作用力)都决定了材料的性质。计算机能够较为精确地处理这样大小的体系。计算机处理化学问题已司空见惯,正如1998年诺贝尔化学奖的颁奖词中所说“化学不再是一门纯实验科学”,因此使用计算机计算单个分子,从而预估该材料的性能是一种可行且成熟的方案。
2012年日本九州大学[8]Adachi课题组研究有机发光二极管(OLED)的发光材料(图2),发现存在着激子自旋统计的限制,即只有25%的单重态激子(S1)参与发光,而剩余的75%三重态激子(T1)不参与。一个合理的方案就是将T1转变为S1,因此理论上就可以将分子设计成明显的电子给体-受体分离结构,这样T1就能够翻越较小的势垒到达S1势能面上。通过设计电子给体(D)为咔唑,电子受体(A)为氰基的分子内电荷转移态(CT)材料,高度密集的给受体组合增加了分子的轨道重叠,使得该分子的最小单三能态的能极差(ΔEST)仅为83 meV,最终该材料的激子利用率高达94%,外量子效率为19.3%。因此,只要理论上能够满足以下两个条件就能获得可能性较高的激子利用率以及发光效率:(1)通过最高占据分子轨道(HOMO)和最低未占分子轨道(LUMO)空间分离的CT材料实现小的S1态和T1态的能级差,在热刺激条件下实现反向系间窜越;(2)利用密集的D-A基团组合,增加轨道重叠的同时增加分子结构刚性,从而抑制分子内的非辐射跃迁,提高辐射发光效率。
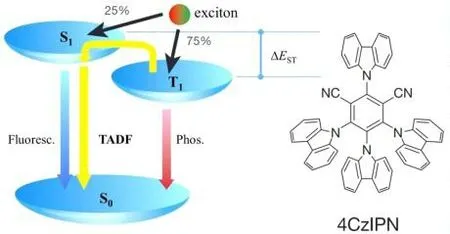
图2 能级示意图和4CzIPN的分子结构Fig. 2 Energy diagram and molecular structures of 4CzIPN
微观上的分子结构的确能够影响材料性能。对于微观上表现较好的分子结构,其最终的材料性能也有可能不是很合适,甚至出现更糟糕的趋势,这些可能与晶体堆积、环境条件变化等相关;但对于微观上表现较差的分子结构而言,其最终性能绝对不可能表现较好。因此分子结构的评估有助于材料的粗筛,减少后续无意义的操作。
2.2 介观之晶体堆积
介观是介于微观与宏观之间的一种体系。分子经过有序组装形成晶体结构,晶体结构同样也能够决定材料的性质。最简单的例子就是石墨和金刚石,两者都是碳的单质,却表现出完全不同的性质,这主要归因于两者的晶体结构的不同,前者是平面层状结构,而后者是立方骨架结构。在中学化学教育中,接触最多的可能是离子晶体,如NaCl、CsCl、SiO2等,呈现出典型的离子晶体的晶胞,但对于有机光电材料而言,由于分析的都是有机晶体,因此晶体呈现的样式也会有所区别。常见有机晶体的分子堆积图案如图3所示[9],其中上为人字形堆积,相邻分子之间不是面对面排列的(如并苯),下为层状图案,二维堆积(如TIPS-并五苯)。
一般情况下,模拟计算微观体系,考虑的都是单分子,且处于真空条件下,而实际体系材料分子均为固相。因此在模拟计算中,常使用晶体堆积的模型来模拟固相环境。固相环境中由于分子受到周围分子的相互作用,因此所处环境与真空有极大差别。具体差异有多大,和分子的柔性、晶体环境中的相互作用非常密切。例如吡咯,属于刚性很强的分子,所以在晶体环境中和在气相中结构差异甚微。而对于柔性体系,晶体环境对构象的影响可能是相当大的,比如二面角能差好几十度。例如联苯这个分子,气相中二面角为41°,中央C与C的键长为0.1479 nm,而在晶体环境中则成了纯平面结构,中央C与C的键长改变为0.1496 nm。此外,在晶体环境中,周围的分子不仅仅影响被考察的分子的几何结构,对其电子结构也同样有影响。因此分子形成的晶体堆积方式对材料的性能也有很明显的影响。

图3 常见有机晶体的两种分子堆积图案Fig. 3 Two molecular packing motifs of common organic crystals
晶体堆积方式对材料性能的影响能有多大?最具有代表性的一个例子是肯塔基大学的Lichtenberger实验组使用UPS光谱测量了一组相似共轭骨架的分子(并五苯和TIPS-并五苯)的气相和凝聚相的IP值(图4)[10]。TIPS-并五苯相比于并五苯,只是其6,13-H被TIPS-基团取代,共轭程度稍微增强,但整体变化不大。从UPS光谱图可知,TIPS-并五苯的气相IP值只比并五苯低0.26 eV,当从气相转移到凝聚相,TIPS-并五苯的IP值变化不大,只有0.4 eV,而并五苯的IP值变化巨大,高达1.7 eV。相比两者的凝聚相IP值,TIPS-并五苯在数值上大于并五苯,逆转了气相下两者的IP值大小顺序,TIPS-并五苯的凝聚相IP较并五苯高达1 eV。比较两者的晶体结构可知,虽然两者都是三斜晶胞,但并五苯晶体呈现人字形堆积,而TIPS-并五苯晶体却是采用砖砌型堆积,如图3所示,因此不同的晶体堆积方式对凝聚相IP值产生了显著的影响,甚至远远超过了强吸、给电子官能团的取代作用(~0.5 eV)。事实上,调节晶体的堆积方式或者给受体分子组成,本质上就是改变分子间相互作用,进而来调控分子晶体材料中的IP和EA值。因此环境(分子聚集和堆积)对材料性能的影响不可忽视。
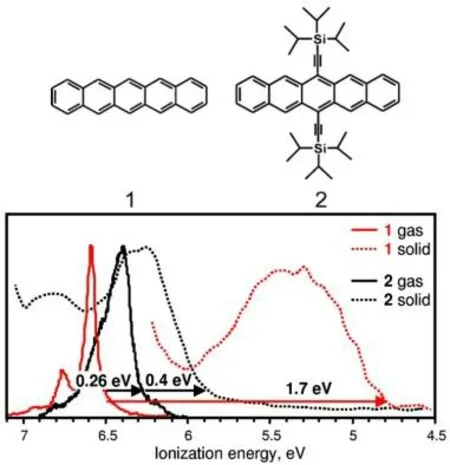
图4 并五苯和TIPS-并五苯的第一电离能带的气相和固相UPS光谱Fig. 4 The gas phase and solid phase UPS spectrum of the first ionization energy band of pentacene and TIPS-pentacene
2.3 宏观之材料表现
除了微观上分子结构和介观上晶体堆积对材料性能产生影响外,在宏观上材料表现和实验工作者的处理方法也同样对材料性能产生很大的影响。一般而言,影响实验结果的因素有:(1)获取数据的方式不同,例如在发射光谱中,使用的是峰值起始值还是峰值最大值[11];(2)考虑是否结构完全弛豫,例如发光材料中,选择的是弛豫前还是弛豫后的状态来观察,使用的是垂直能量还是绝热能量;(3)分子膜结的结晶形态和结晶度,比如晶体不同的堆积方式[12],在界面上呈现出躺式还是站式构型[13];(4)材料表面的性质,需考虑到实验仪器对测量数值的敏感度;(5)不同的基底材料,例如有机太阳能电池中使用的Au,Ag,SiO2或ITO等等,此外表面上的分子取向不同也会影响实验结果;(6)不受控制的环境影响,温度的影响以及晶体/薄膜表面的氧化;(7)仪器的分辨率和一般实验装置。因此,实验上能够最大化的表现出材料的性能也是具有挑战性的课题。
对于中学化学教育而言,材料基因组学看起来“高大上”,但实质上还是“结构决定性质”这个原则的贯彻与实施。材料基因组学充分利用计算机技术与材料科学相结合,压缩现有材料的研发周期,通过新材料研制周期内各个团队的通力合作,注重实验技术、计算技术和数据库之间的协作和共享,降低经济成本和时间成本,为国家的新材料开发、清洁能源和高速信息传递提供保障,提高国家的国际竞争力。
3 虚拟仿真与计算
2012年,杨兵和马於光课题组[14]以三苯胺-菲并咪唑(TPA-PPI)体系为例分析了此类体系的光电特性,利用TPA-PPI作为发射层材料,得到了深蓝色电致发光器件,最大流明效率为5.7 cd/A,外量子效率>5%,单重态激子比例高达28%,超过了自旋统计的上限(25%)。出现这一结果的原因是,可扭转的D-A分子TPA-PPI的发光态激子态能够同时利用局域激发(LE)和电荷转移激发(CT)。分子构型和前线分子轨道的DFT计算分析表明,扭转的D-A构型(20°~55°)是同时包含LE和CT的来源。TPA-PPI在不同扭转角下的基态能量和相应的HOMO和LUMO轨道如图5所示。在90°的扭转角处,可以看到分别在TPA和PPI上明显分离的HOMO和LUMO轨道,这使得TPA-PPI产生了纯CT激发。然而,基于DFT总能量分析,在90°扭转角下的构型不稳定,比扭转角40°的构型能量高了约0.1 eV,而40°扭转角的构型是最低能量构型,其HOMO和LUMO轨道都完全离域到整个TPA-PPI分子上,而不是完全局域在单个的TPA或PPI上。

图5 不同扭转角下的TPA-PPI基态能量(气相)和分子轨道图Fig. 5 The ground-state energy (gas phase) and frontier molecular orbitals (HOMO and LUMO) of TPA-PPI at different twist angles
计算TPA-PPI分子在不同扭转角下的能量,并评估分子最大的能垒大小,使用软件推荐使用Gaussian软件(计算化学领域内被广泛应用的一款综合性量化计算程序包),计算方法为CAM-B3LYP/6-31G(d)。让学生学习分子平衡几何构型优化和限制性优化的相关技术,从不同角度研究一些具体的化学问题,学习通过计算化学手段进行科学研究的办法。
普通高中化学与课程标准[15]明确指出要培养学生宏观辨识与微观操作的核心素养,要求学生能从物质的微观层面理解其组成、结构和性质的联系,形成“结构决定性质,性质决定应用”的观念;能通过观察、辨识一定条件下物质的形态及变化的宏观现象,初步掌握物质及其变化的分类方法,并能运用符号表征物质及其变化;能根据物质的微观结构预测物质在特定条件下可能具有的性质和可能发生的变化;能从宏观和微观相结合的视角分析与解决实际问题。
材料基因组学是覆盖了材料、计算模拟和信息技术的一门新型交叉学科,因此对新材料的最新进展、计算机建模的技巧和信息科学的使用都要较高的要求,这就需要新材料的科研人员、理论与计算模拟工作者以及信息科学的技术人员深入合作。从最开始的单分子或者孤立分子入手,初筛出潜在的高价值体系,再通过分子建模,探索其晶体的堆积方式,进一步细化其研究价值,最后再将其用于实验测量,评估实际的材料性能。材料基因组学的发展,必然极大提高新材料的研发进度,降低材料研发成本,扩展材料学的视野,加强实验与理论研究的合作,为人类社会的发展添砖加瓦。
