E3 泛素连接酶接头蛋白SPOP 抑制前列腺癌的研究进展
2020-12-13综述金晓锋审校
李 倩(综述) 王 健 金晓锋(审校)
(宁波大学医学院生物化学与分子生物系-浙江省病理生理学技术研究重点实验室 宁波 315211)
前列腺癌(prostate cancer,Pca)是第二常见的男性癌症,全世界每年因Pca 死亡25 万例[1],病死率仅次于肺癌[2]。我国Pca 发病率低于西方国家,但随着人口老龄化、饮食和生活习惯改变及血清前列腺特异性抗原(prostate specific antigen,PSA)筛查等体检技术的普及,Pca 发病率和病死率呈逐年上升趋势[3]。
Pca 发病率和死亡率存在显著的种族差异。在不同种族人群中,遗传因素之间的独特相互作用可能导致关键致癌/抑癌基因突变出现差异倾向[4-5]。癌症基因组图谱(The Cancer Genome Atlas,TCGA)发布的数据显示,根据关键致癌/抑癌基因,75%的Pca 可分为7 个分子亚型:ETS家族基因融合(包括ERG、ETV1、ETV4或FLI1 基因),SPOP/CHD1、FOXA1和IDH1基因点突变,另25%的Pca 由其他未知的遗传改变驱动[6-7]。
研究发现,10%~15%的Pca 患者中存在斑点型锌指结构蛋白(speckle-type POZ protein,SPOP)基因的高频突变[8],SPOP突变与TMPRSS2-ETS基因融合互斥,机制可能是SPOP突变后无法降解原本受野生型SPOP 降解的底物,从而抑制TMPRSS2 -ETS融合形成[6,8-13]。这提示SPOP突变可能是早期Pca 发生的驱动事件,对SPOP基因结构和功能的研究有望为Pca 的发生发展、病理分型及分子靶向治疗手段提供新思路。本文将从SPOP基因的结构与功能、Pca 中SPOP的突变情况、SPOP突变介导Pca 发生发展的具体分子机制,以及Pca 临床精准靶向治疗等方面对其研究进展作一综述。
SPOP 蛋白的结构与功能SPOP 蛋白是Cullin3 家族E3 泛素连接酶的接头蛋白,是MATHBTB 核蛋白家族成员之一。SPOP基因位于人染色体17q21.33 上,SPOP 蛋白由374 个氨基酸残基组成,相对分子质量为42 000,主要结构域(图1)包括N 端的底物结合结构域MATH(31~164 位氨基酸残基)、与Cullin3 相互作用的BTB 结构域(184~297位氨基酸残基)、3-box(300~327 位氨基酸残基)和C 末端的核定位(NLS)序列(365~374 位氨基酸残基)[14-15]。
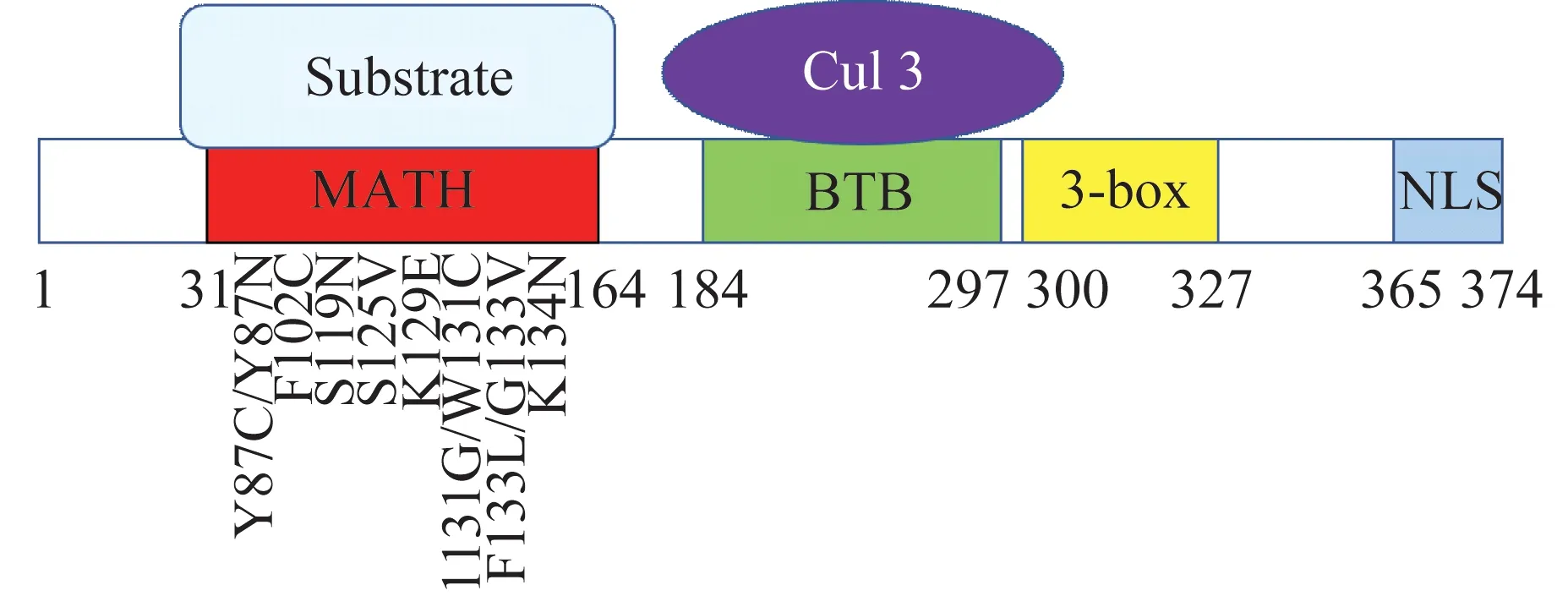
图1 SPOP 蛋白的结构及Pca中SPOP 基因高频突变位点的示意图Fig 1 The diagram of SPOP protein and the distribution of most comman mutation in the SPOP gene found in prostate
晶体学和小角度X 射线散射分析(small angle X-ray scattering,SAXS)数据表明,SPOP 结构最显著的特征是MATH 结构域连接BTB/3-box 结构域,生成由2 个底物结合位点和2 个催化核心组成的二聚体泛素连接酶[15]。BTB 和3-box 结构域的协同二聚作用促进了线性的SPOP 二聚形成。由于这种特殊的线性二聚结构,SPOP 结构域可以募集底物并延长泛素链,使其灵活变动方向,从而获得更高的亲和力并和更多的构象选择来介导泛素化。SPOP 通过MATH 结构域选择性地募集底物,而其BTB 和3-box 结构域介导寡聚化,并与Cullin3 相互作用[16],进而泛素化修饰底物,调控底物的蛋白质水平(泛素化降解)或者功能活性(非降解型泛素化修饰,图2A)。研究发现SPOP 的底物参与各种基本的细胞功能活动,例如死亡结构域相关蛋白(death domain associate protein 6,Daxx)参与细胞转录、细胞周期和凋亡[17];雄激素受体(androgen receptor,AR)参与细胞的信号转导[18];内质网(endiplasmic reticulum,ER)应激反应转录因子(DNA damage inducible transcript 3,DDIT3)在内质网应激时转入细胞核内,调控凋亡基因的表达,维持细胞稳态[19]。因此正常功能的SPOP 在细胞周期、信号转导和维持细胞功能稳态等过程中均起重要作用[20-22]。
2010 年在一项关于58 种肿瘤体细胞突变的研究发现,SPOP是Pca 的高频突变基因[23],且突变位点几乎都集中于MATH 结构域。此后,大量研究发现Pca 来源的SPOP突变体通过两种方式影响底物蛋白的功能。其一,突变的SPOP丧失结合底物的能力,使得促细胞生长的底物(如AR 蛋白)水平升高从而诱发Pca[24](图2C)。其二,野生型SPOP通常通过二聚体形式起作用,如果存在SPOP突变体,会影响正常二聚体的功能,进而破坏野生型SPOP 的蛋白功能(显性负效应),例如SPOP突变体通过显性负效应增加脊椎动物formin 蛋白(inverted formin-2,INF2)在内质网中的定位,促进线粒体分裂,这种线粒体分裂参与Pca 细胞的迁移和侵袭(图2B),从而诱发Pca[25]。高通量测序发现,Pca 患者体内SPOP 的相关底物蛋白质存在突变,这类突变使得底物无法与SPOP 结合,逃逸了被泛素化修饰降解的命运,从而诱发Pca(图2D)。完整梳理SPOP突变如何影响正常SPOP 的结构和功能,以及如何影响底物的功能,是对SPOP突变的Pca 患者实行精准治疗的前提。
Pca 中SPOP 基因突变的突变率和突变位点1997 年Nagai 等[26]首次发现SPOP,因其含有1 个POZ 结构域和在核内呈斑点状散在分布的特征而命名为斑点型POZ 蛋白。随后的研究阐明了SPOP 作为E3 泛素连接酶衔接蛋白的功能[27-28]。2012 年Barbieri 等[8]首次证实SPOP突变与Pca 发生发展密切相关。
多项全基因组测序或外显子测序研究发现,SPOP突变在原发性Pca 中占6%~13%,在转移性Pca 中占14.5%,不同种族和民族背景下的Pca 患者中SPOP的总体突变率为4.6%~14.4%[7]。
Pca 中已发现的SPOP突变均发生在与底物结合的MATH 结构域,这些突变显著降低了SPOP 与底物结合的亲和力,导致SPOP 介导的泛素化功能失活(图2B 和2C)。通过二代测序(next generation sequencing,NGS)检测,Pca 中SPOP基因高频突变位点包括Y87C、Y87N、F102C、S119N、F125V、K129E、W131C、W131G、F133L、F133V 和K134N[8]。其中,F133 发生频率最高(约50%),其次是Y87、W131、F102、F125、K129、K134 和S119[9](图1)。SPOP基因的突变聚集在一个约200 bp 的区域内,为常规DNA 诊断提供了新的检测靶点。Barbier等[8]发现,在大多数Pca 中只有1 个拷贝的SPOP等位基因发生突变,并且SPOP突变体以显性负效应抑制野生型SPOP,从而发挥其肿瘤促进功能。同时,底物蛋白必须具备特异的SBC 基序(SPOPbinding consensus motif,SBC motif),即SPOP 结合的共有基序符合Ω-π-S-S/T-S/T(Ω:非极性氨基酸,π:极性氨基酸,S:丝氨酸,T:苏氨酸)的氨基酸序列[15]。该基序始终存在于SPOP 降解的底物蛋白中,包括磷酸酶Puc[29]、转录调节因子Ci/Gli[30]和Daxx[17]和染色质组分MacroH2A[27]等。AR 的SBC基序在部分Pca 患者体内存在突变,无法被SPOP识别,逃避了SPOP 介导的泛素化降解,从而促进Pca 发生[24](图2D)。
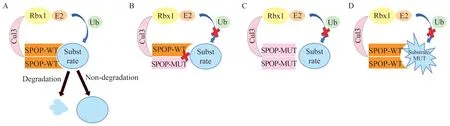
图2 SPOP 野生型及突变体与底物结合的示意图Fig 2 The diagram of the interaction between SPOP-WT/MUT and substrate
SPOP 影响Pca 发生发展的分子机制SPOP蛋白在Pca 细胞中起着关键性的抑制作用[31],Daxx[17]、AR[18]、DDIT3[19]、类固醇受体共激活因子3(steroid receptor coactivator 3,SRC-3)[32]、ETS 相关基因(ETS-related gene,ERG)[33]、DEK[34]、细胞性骨髓细胞瘤病毒癌基因(cellular-myelocytomatosis viral oncogene,c-MYC)[35]、细胞周期蛋白20(cell division cycle,CDC20)[31]、egl-9 家族缺氧诱导因子2(egl-9 family hypoxia inducible factor 2,EglN2)[36]、溴结构域家族蛋白4(bromodomain-containing protein 4,BRD4)[37]、组蛋白去乙酰化酶(histone deacetylase 6,HDAC6)[38]和程序性死亡受体-配体1(programmed death-ligand 1,PD-L1)[39]等均被证实为SPOP 的底物,并且由于SPOP突变亚型失去结合能力,这些底物不能被泛素化降解,导致蛋白功能紊乱而诱发Pca。突变型SPOP 影响Pca 发生发展产生的机制:介导AR 依赖的信号通路,DNA 损伤修复,肿瘤的免疫反应及线粒体动态调节和内质网应激等(图3)。
SPOP 与AR 依赖性通路AR 是关键的转录因子,对于正常前列腺细胞的生长和存活至关重要[18]。SPOP 以AR 依赖性方式促进AR 蛋白的泛素化和蛋白酶体降解,并抑制AR 介导的Pca 细胞的生长[24]。AR 的可变剪接是其中一种重要机制,AR 的铰链结构域中具有SBC 基序645ASSTT649。失去铰链域的AR 剪接体不能被SPOP 识别,逃避了SPOP 介导的泛素化降解,从而促进Pca 发生。目前发现的Pca 相关SPOP突变体主要集中于MATH 结构域,均失去了结合并泛素化降解AR 的能力。雄激素减弱SPOP 结合并降解AR 的能力,而抗雄激素会增强这种能力。一种可能的解释是,雄激素与AR 结合后改变了其构象,从而影响其SBC 基序与SPOP 结合;而抗雄激素阻断雄激素与AR 的结合,间接促进SPOP 与AR 结合,有效抑制AR 信号通路[24]。
SRC-3 是SPOP 的靶向底物,也是AR 的激活蛋白。野生型SPOP 能结合并泛素化降解SRC-3,而Pca 来源的SPOP突变体失去结合SRC-3 的能力,SRC-3 持续性激活AR 信号通路而诱发Pca[31-32]。
SRC-3 通过胰岛素样生长因子1(inslin-like growth factor 1,IGF-1)的转录上调激活磷脂酰肌醇3-激酶/哺乳动物雷帕霉素蛋白(phosphoinositide 3-kinase/mammalian target of rapamycin,PI3K/mTOR)信号转导[40]。在SPOP 突变型Pca 中没有富集AR 信号通路相关的基因,而是富集PI3K/mTOR 信号转导相关的基因,因此提出PI3K 和AR信号通路存在显著的负反馈作用,即PI3K 激活导致AR 信号下调[41-44]。而SPOP突变体在体内外通过SRC-3 激活PI3K/mTOR 信号转导,并上调AR 相关转录因子和共激活因子的网络,同时维持AR 活动并对抗PI3K 介导的反馈抑制,从而有效激活并协调这两条对Pca 发生发展至关重要的途径[45]。
ERG 癌蛋白也是SPOP 的重要底物。SPOP 可调节ERG 蛋白水平,并且SPOP突变会导致ERG的累积,从而促进癌细胞侵袭与增殖[46]。2013 年Chen 等[47]提出ERG 在AR 信号转导中的“先驱因子”作用,即维持AR 转录水平。2015 年再次证明ERG基因融合形成的截短ERG 体不受SPOP 的调控,SPOP突变诱导的癌症表型很大程度上是通过ERG 介导的。所有TMPRSS2-ERG 融合转录本始终受AR 正调控和SPOP 负调控[24,48]。克服截短体ERG 对泛素降解的抵抗,有望成为Pca 的治疗新靶点。
SPOP 与DNA 损伤修复基因组不稳定是人类癌症的基本特征之一。与其他肿瘤亚型相比,Pca 中SPOP突变亚型的基因组重排数量高,这表明SPOP突变亚型肿瘤具有高度的基因组不稳定性[49]。为维持基因组的稳定性,机体进化出高度保守的应答体系——DNA 损伤应答(DNA damage response,DDR)。DDR 包含4 条子途径:DNA 损伤修复、DNA 损伤检查点、转录反应和凋亡[50]。
SPOP 是Pca 细胞中DDR 的参与者,提示SPOP 对维持基因组稳定性和DDR 完整性具有关键作用[49,51]。Boysen 等[49]研究发现,SPOP 可通过调节DNA 双链断裂(DNA double-strand breaks,DSB,DSB)修复来维持基因组稳定性。SPOP突变改变了DNA 修复过程,削弱了同源重组(homologous recombination,HR),促进易出错的非同源末端连接(non-homologous end joining,NHEJ)而导致细胞基因组不稳定,进而诱发Pca[49]。Hjorth-Jensen 等[52]发现SPOP 在抵抗复制压力过程中起到重要作用。野生型SPOP 与多数参与转录、mRNA 剪接和出核的蛋白质有关,例如BRCA2、ATR、CHK1 和RAD51 等。SPOP 促进这些复制因子的转录表达,起到减少复制压力的作用。因此,SPOP 功能的丧失会促进自发复制压力和基因组不稳定,尤其是SPOP突变或敲低后抑制RAD51 基因的形成,可引起自发复制压力、损伤修复缺陷和异常的细胞周期[52]。SPOP突变引起修复受损并导致细胞对电离辐射(ionizing radiation,IR)过敏,进一步影响DNA 损伤检查点,并诱导细胞凋亡。SPOP促细胞凋亡的机制可能是BTB/POZ 结构域影响转录调节因子锌指蛋白的折叠,并调节诱导凋亡的基因转录,具体机制有待进一步研究。
AR 信号通路也具有调节DDR 的功能。抑制AR 信号可使PCa 细胞对IR 敏感,并且涉及一些DNA 修复基因的表达,进一步提示SPOP 在维持基因组稳定中的重要作用[18,53]。
SPOP 与免疫近年来肿瘤免疫生物治疗已成为治疗Pca 的突破点[54]。机体免疫系统不仅免疫监视并清除肿瘤细胞,还能促进肿瘤免疫逃逸。
先天信号转导者髓系分化初级反应蛋白88(myeloid differentiation factor 88,MyD88)是Toll 样受体信号通路中的重要转导蛋白,其依赖的信号通路以及调控的基因产物在固有免疫和适应性免疫中均发挥关键作用。Guillamot 等[55]通过蛋白组学研究发现,SPOP 泛素化修饰MyD88,并促进其进入蛋白酶体途径降解,从而调控紧急造血程序向稳态造血程序的转换,以限制全身炎症反应发生。SPOP突变时,MyD88 不能被正常降解,导致IRAK4 激酶过度磷酸化,异常激活MyD88-IRAK4下游的炎症反应因子NF-κB 和AP-1,从而促进肿瘤细胞的侵袭和转移[55]。Jin 等[56]进一步研究了SPOP 与MyD88 的互作机制,发现在淋巴瘤中SPOP 通过非降解型泛素化修饰MyD88 差异可能由于细胞不一致引起。两项研究均表明,SPOP 阻断了MyD88 小体(Myddosome)的组装和下游NFκB 的激活,证明了SPOP 调控MyD88 对于免疫应答的作用[56]。
研究显示程序性死亡受体1(programmed death 1,PD-1)/PD-1 配体(PD-1 ligand,PD-L1)在许多恶性肿瘤中高表达,PD-1/PD-L1 信号通路的激活可形成免疫抑制性肿瘤微环境,造成肿瘤免疫逃逸,导致肿瘤发生发展[57]。具体机制为:PD-1 及PD-L1在炎症反应时抑制周围组织T 细胞的活性,并通过诱导活化的T 细胞凋亡、促进T 细胞衰竭、增强调节性T 细胞的免疫抑制功能、抑制T 细胞的增殖与活化和产生IL-2 等方式调控自身免疫,同时介导肿瘤的免疫逃逸[58]。Zhang 等[39]发现,PD-L1 被SPOP介导的泛素化修饰途径所降解,在该途径中SPOP受到细胞周期调节蛋白Cyclin D-激酶CDK4 对其磷酸化的影响。SPOP突变后PD-L1 降解受抑制,导致PD-L1 水平升高以及肿瘤浸润淋巴细胞数量减少。动物实验发现,联合使用CDK4/6 抑制剂和抗PD-1 治疗后,肿瘤浸润淋巴细胞恢复到正常水平,显著抑制肿瘤进程,并提高小鼠的生存率[39]。联合使用CDK4 抑制剂和PD-1/PD-L1 阻滞剂具有治疗肿瘤的作用。了解SPOP 与免疫相关的信号通路对于临床免疫治疗靶点的研究具有重要意义。
SPOP 与其他通路
SPOP 与线粒体裂变 越来越多的证据表明线粒体的裂变和融合在调节细胞运动、迁移和侵袭中起积极作用[59-60]。线粒体裂变的关键参与者反向蛋白INF2 是SPOP 的泛素化底物之一,但SPOP 的泛素化修饰并不会降解INF2,而会减少ER 中INF2定位以及与线粒体相关的DRP1 斑点形成,消除其促进线粒体分裂的能力。Pca 相关的SPOP突变体失去泛素化修饰INF2 的能力,继而INF2 持续促进线粒体分裂,诱导Pca 细胞迁移和侵袭[25]。
SPOP 与内质网应激 ER 功能被破坏后会产生错误折叠和未折叠的蛋白质积聚,未折叠的蛋白反应会减轻这种应激压力并恢复ER 稳态,从而促进细胞存活和适应,如果不能及时缓解压力,则会触发凋亡性细胞死亡[61]。在ER 应激触发的凋亡中,DDIT3 被强烈诱导,转移至细胞核,抑制抗细胞凋亡基因的表达,并激活促凋亡基因表达[62]。SPOP识别DDIT3 并对其进行泛素化,促进蛋白酶体途径降解,而突变体丧失此功能,不能抑制ER 应激诱导的细胞凋亡,进而推动癌症发展[19]。SPOP突变的肿瘤可能对内质网应激药物更为敏感,因为这些肿瘤在调节DDIT3 蛋白更新方面存在缺陷。

图3 SPOP 影响Pca 发生发展的分子机制Fig 3 Schematic of the proposed mechanism through which SPOP suppresses prostate cancer
Pca 组织中SPOP 蛋白表达的临床意义
PROTAC 技术用于Pca 治疗 Pca 发生发展的遗传因素复杂多样,有显著的肿瘤异质性,在基因组序列、表观遗传学等分子水平上存在明显差异[63]。因此,寻找Pca 不同分子分型的差异靶点对肿瘤的精准治疗十分重要。针对SPOP突变的分子亚型对于Pca 发生发展的影响,临床上已研制出多种不同机制的治疗药物。
SPOP突变的患者体内AR 蛋白水平处于较高水平,AR 在Pca 的发展,特别是在去势抵抗性Pca中起关键作用。对于这类患者至关重要的是重新激活E3 泛素降解系统,有效降低AR。目前临床上使用的AR 抑制剂以恩杂鲁胺(enzalutamide)为主,需要维持恩杂鲁胺高体内浓度,且该抑制剂在AR蛋白高表达时往往失去活性[64]。由于目前无法通过药物靶向突变体SPOP 使其恢复野生型活性,考虑到SPOP 在肿瘤组织中的失活突变导致促癌蛋白AR 的积累,降低这些促癌蛋白的蛋白水平应当可以抑制SPOP突变型Pca 的恶性增殖。蛋白质降解靶 向 联 合 体(proteolysis targeting chimeras,PROTAC)技术是靶向降解特异蛋白质的一种新方法,PROTAC 是一种双功能杂合分子,可同时结合E3 泛素连接酶和靶蛋白,从而驱动靶蛋白被泛素结合进入蛋白酶体途径而降解[65](图4)。考虑到SPOP突变患者体内的AR 无法被降解,Jemilat等[66]设计了特异靶向AR 的PROTAC——ARCC-4,其降解AR 蛋白的效率比恩杂鲁胺强约10 倍。ARCC-4 以低浓度有效降解与抗雄激素治疗相关的AR 突变体,并在AR 蛋白高水平中保持其降解AR和抑制细胞增殖的能力,但ARCC-4 的细胞通透性及其差向异构体的效价远低于恩杂鲁胺[66]。经过改进,ARV-110 在保证低浓度高效靶向降解AR 的基础上解决了细胞通透性差的问题,并获得临床药物试验许可。2020 年5 月的临床实验结果显示,ARV-110 在患者体内能成功降解AR[67]。

图4 利用PTOTAC 技术降解AR 蛋白的模式图Fig 4 The diagram of degradation of AR using PROTAC technique
含溴结构域和ET 域(bromodomain and extraterminal,BET)蛋白也是Pca 的治疗靶点,BET 抑制剂(如JQ1 和I-BET)已广泛用于临床治疗,一方面降低表观遗传调节剂BET 蛋白以抑制细胞分裂[68-70],另一方面通过阻断BRD4 而抑制AR 介导的转录。Zhang 等[37]发现野生型SPOP 能够结合BRD4,促进其进入蛋白酶体途径降解。然而,在部分SPOP突变的Pca 患者中BRD4 无法被正常降解,进一步导致SPOP突变的PCa 亚型中AKTmTORC1 信号的激活和对BET 抑制剂的抗性[45,71-72],对该类抑制剂产生耐受性。因此,BET 抑制剂仅适用于不存在SPOP突变或存在SPOP突变但不产生耐药性的患者,这为Pca 的精准治疗提供了新思路。考虑到SPOP突变的Pca 患者体内缺乏正常活性的E3,靶向BET 的PROTAC 降解剂ARV-771 对于临床上具有BET 抑制剂抗性的Pca患者具有治疗潜力[73]。
免疫治疗 免疫疗法已成为广泛研究的癌症治疗方法。免疫检查点抑制剂阻断肿瘤细胞和/或免疫细胞的凋亡信号蛋白,以防止肿瘤细胞诱导免疫细胞死亡。Zhang 等[39]研究表明,CDK4/6 抑制剂联合PD-1/PD-L1 阻滞剂具有抑制肿瘤免疫侵袭的作用。CDK4/6 抑制剂能通过抑制SPOP 磷酸化使其被E3 连接酶APC/C 的共激活因子Cdh1 降解,从而在PD-L1 蛋白水平升高的基础上使用PD-1/PDL1 阻滞剂达到治疗目的。
结语SPOP 介导Pca 发生发展的机制有待进一步探索,新的分子靶点有望成为Pca 新的生物学分型标志物,并为Pca 患者个体化治疗、基因治疗药物研制以及临床用药等提供新的思路。
