铁死亡的发生机制及其在血液系统肿瘤中的研究进展
2020-12-09殷贤青刘容容
殷贤青,刘容容
(广西医科大学第一附属医院血液内科,南宁 530000)
细胞死亡是维持个体正常生长发育及内环境稳态的重要环节之一,经典的细胞死亡模式为凋亡、自噬和坏死。 铁死亡是一种新的细胞死亡调控形式,其形态学特点是细胞体积缩小,线粒体膜密度增加,嵴减少,它在机制上不同于细胞坏死和凋亡,主要特点是活性氧(ROS)的生成、脂质过氧化和铁的积累。随着近年来铁死亡机制研究的深入,许多研究表明铁死亡与肿瘤的发生发展密切相关。白血病和淋巴瘤是血液系统恶性肿瘤,其现有的治疗方案治疗有效率低,仍需探索新的治疗模式,而铁死亡的研究进展也为血液系统肿瘤的治疗提供了新的思路。
1 概述
铁死亡是Dixon等[1]在2012年提出的由erastin和RSL3等小分子诱导的依赖铁的一种新的死亡模式,主要特点是活性氧(ROS)的生成、脂质过氧化和铁的积累。特定的小分子化合物作用于细胞内特异性靶点,引起抗氧化剂谷胱甘肽(GSH)或谷胱甘肽过氧化物酶4(GPX4)减少,导致细胞内ROS堆积,细胞发生脂质过氧化,在铁协同作用下诱发细胞铁死亡[2-3]。游离胱氨酸经胱氨酸谷氨酸转运受体(systemXc-)转运进细胞内,为合成GSH的底物,谷胱甘肽是一种主要的氧化还原分子,其功能是通过将一个电子提供给谷胱甘肽过氧化物酶4 (GPX4)来防止铁中毒[4],而GPX4是唯一能减少磷脂过氧化氢的酶[5](图1)。但上述通路引起的脂质过氧化水平增高通过什么机制引起铁死亡仍是一个未解之谜。
白血病和淋巴瘤是最常见的血液系统恶性肿瘤,主要治疗方法是化疗和干细胞移植,虽然干细胞移植治疗水平在近年来有较大提升,但其应用具有一定的限制性。而化疗方案的缓解率不高,且近年来并没有实质性的进展。因此仍需探究对患者受益较大的治疗方案。铁死亡作为细胞死亡模式之一,是肿瘤研究和治疗中的热门研究方向[6-7]。目前铁死亡在血液系统肿瘤中研究较多的为通过调节铁死亡的调节因子,影响细胞对铁死亡的敏感性,从而调节肿瘤细胞的铁死亡水平。
2 铁死亡机制及在血液系统肿瘤中的研究
2.1 脂质过氧化与血液系统肿瘤
活性氧(ROS)由正常生理过程产生的,在细胞信号转导和组织稳态中起重要作用。然而,过量的活性氧自由基对细胞成分产生不利的修饰,如脂质、蛋白质和DNA损伤[8-9]。由于生物细胞膜或细胞器膜富含高多不饱和脂肪酸(PUFAs),特别容易受到ROS损伤,这被称为“脂质过氧化”,ROS诱导的脂质过氧化在细胞死亡中起着重要的作用。脂质过氧化直接损伤磷脂,也可作为诱导细胞程序性死亡的细胞死亡信号[10-11]。在erastin和RSL3诱导的铁死亡中,均伴有ROS的累积,通过高通量筛选发现ferrostatin-1 (Fer-1)和 liproxstatin-1 (Lip-1)可阻止erastin诱导的ROS累积,从而特异性的抑制RSL诱导的铁死亡,这也验证了ROS累积对促进铁死亡的重要作用[1,12]。
GPX4是一种抗氧化酶,GPXs家族成员之一,也是人类基因组17中含有硒半胱氨酸的25种蛋白质之一,它以谷胱甘肽为辅助因子,将脂质过氧化物还原为脂质醇,这一过程阻止了活性氧ROS的合成[13-14]。GPX4是RSL3的蛋白靶点,RSL3能特异性抑制GPX4活性,导致细胞内ROS堆积,从而诱导细胞的铁死亡[15],而过表达的GPX4可降低铁死亡水平[16]。Pedro等[17]人证明敲除GPX4基因会导致细胞发生铁死亡。硒可以通过转录因子TFAP2c和Sp1的协同激活来增强GPX4和这个转录程序中的其他基因,有效地抑制GPX4依赖的铁死亡,而硒的缺失会使GPX4失去活性,使细胞对氧化损伤的敏感性增高[18-19]。此外,Fino2和FIN56可通过间接抑制GPX4的水平和活性来诱导铁死亡,而不影响谷胱甘肽的水平[20-21]。
胱氨酸谷氨酸转运受体(systemXc-)是细胞膜转运体的一个组成部分,由SLC7A11和SLC3A2组成的异质二聚体,负责胞外胱氨酸和胞内谷氨酸的交换[22]。在生理状态下,细胞外的胱氨酸通过systemXc-转运进细胞内,为合成抗氧化剂谷胱甘肽的底物,而谷胱甘肽是清除活性氧的主要成分[23]。阻断systemXc-会抑制半胱氨酸依赖的谷胱甘肽(GSH)合成,进而损害细胞的抗氧化防御能力,从而促进ROS的积累,诱导细胞发生铁死亡。Wang等[24]人发现敲除Slc7a11基因小鼠体内胱氨酸/半胱氨酸的来源降低,从而限制了随后的GSH合成,增加了细胞对铁超载诱导的铁死亡的敏感性。而b-巯基乙醇(b-ME)可以通过另一种途径促进胱氨酸的摄取,从而绕过对systemXc-的抑制,它可以强烈地抑制由谷氨酸诱导的HT-1080细胞的死亡[1]。
综上,铁死亡过程中伴随着ROS的积累和脂质过氧化水平的增高,而GPX4通过抗氧化减少细胞内ROS累积来减轻细胞对铁死亡的敏感性,GPX4的水平和活性能影响铁死亡水平。此外,systemXc-通过影响GSH的代谢调节ROS的平衡,为参与铁死亡的重要一环。
Probst等[25]人使用急性淋巴细胞白血病(ALL)细胞系为细胞模型,RSL3处理后该细胞系细胞死亡,这种死亡过程伴随着脂质过氧化增加水平的增高,加入脂质过氧化抑制剂Fer-1或脂氧合酶(LOX)后可抑制这种细胞死亡,且铁螯合剂DFO可逆转RSL3触发的细胞死亡,这些结果表明ALL细胞对RSL3诱导的铁死亡敏感。此外,Yang等[13]人通过检测117个来自不同组织的癌细胞系对erastin敏感性,数据分析结果显示弥漫性大B淋巴瘤(DLBCLs)特别敏感,且在经erastin处理后的DLBCL细胞系中产生了脂质过氧化物,而使用亲脂性抗氧化剂可以挽救细胞死亡,表明在在此细胞系中发生的细胞死亡具有铁死亡的特征。进一步分析了DLBCL细胞系和其他造血细胞系对203种不同致死化合物的敏感性,发现DLBCL细胞系对所有化合物的平均耐药能力较弱,表明DLBCLs对erastin诱导的ferroptosis的敏感性增强并不是由于对所有化合物都具有普遍的敏感性。上述研究表明,白血病和淋巴瘤细胞对铁死亡敏感性高,且伴随ROS堆积过多、脂质过氧化水平增高的现象,与目前所知的铁死亡经典通路相符合。
近年来有学者发现急性髓系白血病(AML)和Burkitt淋巴瘤对可促进铁死亡的化合物敏感。酪啡肽(Typhaneoside, TYP)是蒲黄花粉提取物中的主要黄酮类化合物,处理AML细胞后通过促进AMP激活蛋白激酶(AMPK)信号的激活,显著地触发AML细胞的自噬,最终导致铁蛋白降解、ROS积累,同时伴有线粒体功能障碍,最终导致细胞铁死亡[26]。此外,Wang等[27]人研究了青蒿琥酯对Burkitt淋巴瘤细胞基因表达及细胞抑制的影响,结果显示青蒿琥酯可诱导Burkitt淋巴瘤细胞的铁死亡,从而导致内质网应激反应,激活ATF4-CHOP-CHAC1通路,并降解了细胞内的GSH,从而削弱了淋巴瘤细胞对铁死亡的抵抗能力,增强了Burkitt淋巴瘤细胞的铁死亡,这可由Lip-1、Fer-1和DFO对细胞的保护作用得到证明。这些研究为促铁死亡化合物在血液肿瘤治疗领域的应用提供了新思路。
人淋巴瘤细胞和白血病细胞不能将蛋氨酸转化成胱氨酸,因此,细胞外胱氨酸摄取是其生长和进展所必需的。有趣的是,与其他系统实体瘤中高表达的SLC7A11水平相比,在慢性淋巴细胞白血病(CLL)中SLC7A11表达下调,systemXc-转运胱氨酸能力下降,可促进细胞内ROS增加,这表明了CLL与铁死亡密切相关[28]。
除了体外研究外,有临床研究结果显示[29],在DLBCLs患者中,GPX4的表达率为35.5%(33/93),GPX4阳性组的总生存期和疾病无进展生存期较GPX4阴性组差,这可能与GPX4能减少细胞内脂质过氧化水平而降低细胞对铁死亡的敏感性的有关。
综上,细胞发生铁死亡过程中伴随着ROS的积累和脂质过氧化为铁死亡发生机制的重要环节,对GPX4和systemXc-等通路的调节最终均通过影响细胞内ROS的稳态来改变细胞对铁死亡的敏感性。研究表明,通过多通路调节增加细胞内ROS的堆积可增加白血病及淋巴瘤细胞对铁死亡的敏感性,从而为临床上治疗血液系统肿瘤的药物选择提供了新的研究方向。
2.2 p53与血液系统肿瘤
人们普遍认为p53介导的细胞周期阻滞、凋亡和衰老是抑制肿瘤的主要机制。Jiang等[30]人报道,p53通过抑制SLC7A11的转录降低了胱氨酸的摄取,减少细胞内GSH,增加细胞内ROS堆积,从而增加细胞铁死亡的易感性。他们使用了p53的一种不能诱导其它形式的细胞凋亡,但保留了调节SLC7A11表达的能力的乙酰化缺陷突变体p533KR,发现SLC7A11在许多类型的人类癌症中过表达,而高水平的SLC7A11表达可以显著消除p533KR诱导的肿瘤生长抑制活性,表明这种抑制活性与细胞周期阻滞、凋亡和衰老无关。同时,他们发现高水平的活性氧可以触发p53介导的铁死亡。而p53对细胞ROS水平的调节作用是一个有趣的过程。在细胞内为低水平或基础ROS水平时,p53可以阻止细胞积累致命水平的ROS,然而,当ROS水平异常升高时,p53可能会通过铁死亡来促进细胞的清除。由此可知,p53可以通过影响细胞内ROS水平来调节细胞铁死亡水平。
ALOX12基因位于人类染色体17p13.1上,位置非常接近TP53位点,部分学者认为许多人类肿瘤已经丢失了一个ALOX12等位基因[31]。Chu等[32]人采用分别缺失脂氧合酶六种亚型的p533KRH1299细胞,经过氧化叔丁醇(TBH)处理后检测ROS诱导的铁死亡水平,发现ALOX12的功能缺失特异阻断了p53介导铁死亡,且SLC7A11通过特异性结合ALOX12来抑制其酶活性,从而证实p53可以通过抑制SLC7A11的转录从而抑制systemXc-系统,间接激活ALOX12脂氧合酶活性,导致ROS诱导下 ALOX12依赖性的铁死亡,而这条诱导铁死亡的途径独立于GPX4途径。因此,p53可以通过调节SLC7A11的转录水平和活性来调控铁死亡水平。进一步研究发现,ALOX12缺失可抑制p53介导的p53型铁死亡并加速myc诱导的肿瘤发生,在Eμ-Myc老鼠模型中,损失一个Trp53等位基因时会显著加速Myc诱发的经典Eμ-Myc淋巴瘤模型的形成,而丢失一个ALOX12等位基因会缩短这类小鼠的中位生存期。
综上,p53通过调节ROS水平增加细胞随铁死亡的敏感性,而p53功能性缺失对Eμ-Myc淋巴瘤的发生和预后有重要作用。
2.3 铁代谢与血液系统肿瘤
铁是人体内参与多种代谢的重要金属离子之一,对促进铁死亡的发生起着重要作用。铁离子参与铁死亡的具体机制尚不明确,研究表明,在生理条件下,循环游离铁与转铁蛋白结合,体内铁代谢维持稳态,当体内出现铁超载时,血清转铁蛋白结合接近饱和,循环中存在非转铁蛋白结合铁(NTBI)[4,33-34],而过量的铁可通过Fenton反应使细胞内ROS含量升高,从而促进铁死亡[35]。使用枸橼酸铁(FAC)在体外处理小鼠原代肝细胞和骨髓来源巨噬细胞(BMDMs) 显著增加了细胞的脂质过氧化水平,降低了NADPH含量和细胞生存能力,而铁死亡抑制剂和铁螯合剂可逆转上述改变[24]。此外该团队还发现,Slc7a11-/-小鼠模型在基础铁条件下不发生铁死亡,高铁饮食引起小鼠体内非转铁蛋白结合铁增高,GSH水平降低, ROS水平升高,表明,铁诱导的铁死亡是一个不同于erastin诱导的铁死亡的过程。
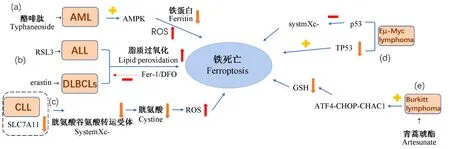
注:(a):酪啡肽处理AML细胞后可通过激活AMPK信号导致AML细胞铁死亡,并伴随铁蛋白降解及ROS累积;(b):RSL3、erastin处理ALL细胞、DLBCLs后致细胞发生铁死亡,伴随脂质过氧化水平增高,而抗氧化剂及DFO可抑制其发生;(c):CLL细胞中SLC7A11表达下调,systemXc-转运胱氨酸能力下降,致细胞内ROS增加,促进细胞铁死亡;(d):p53可抑制systemXc-而促进Eμ-Myc淋巴瘤细胞发生铁死亡,而缺失TP53基因会加速Eμ-Myc淋巴瘤模型形成;(e):青蒿琥酯可通过激活ATF4-CHOP-CHAC1通路并降解GSH诱导Burkitt淋巴瘤细胞铁死亡。图1 铁死亡与血液系统肿瘤Note. (a), Typhaneoside treatment of AML cells resulted in ferroptosis in AML cells by activating AMPK signaling, accompanied by ferritin degradation and ROS accumulation. (b), RSL3 and erastin treatment of ALL and DLBCLs resulted in ferroptosis, accompanied by increased lipid peroxidation, which was inhibited by antioxidant and DFO. (c), Expression of SLC7A11 in CLL cells was down-regulated, and the systemXc- transporter cystine ability was decreased, leading to the increase of intracellular ROS and the promotion of cell ferroptosis. (d), p53 inhibits systemXc- and promotes ferroptosis in Eμ-Myc lymphoma cells, while the deletion of TP53 gene accelerates the formation of Eμ-Myc lymphoma model. (e), Artesunate can induce ferroptosis in Burkitt lymphoma cells by activating ATF4-CHOP-CHAC1 pathway and degrading GSH.Figure 1 Ferroptosis and hematological malignant tumor
此外还有许多研究结果与其相一致。核受体辅激活因子4 (NCOA4)是铁蛋白在铁死亡中的选择性自噬翻转的选择性受体,Hou等[36]人在PANC1细胞中敲除NCOA4,细胞内二价铁水平减低,减少了erastin诱导的铁死亡,而通过转染过表达NCOA4的细胞中二价铁水平增高,且铁死亡水平增高,因此,NCOA4介导的铁蛋白降解导致的细胞内铁离子增加参与了铁死亡。GPX4抑制剂处理的细胞分泌的大量外泌体中含有铁蛋白,在发生铁死亡时,prominin 2水平与细胞内游离铁水平呈负相关,表明外泌体将细胞内铁排出细胞能保护细胞免于发生铁死亡[37]。
近年来有学者发现急性髓系白血病(AML)和Burkitt淋巴瘤对可促进铁死亡的化合物敏感。双氢青蒿素(DHA) 在G0/G1期强烈抑制AML细胞系的活力并阻滞细胞周期,进一步研究发现,DHA通过调控AMPK/mTOR/p70S6k信号通路的活性诱导自噬,加速铁蛋白的降解,增加不稳定的铁池,促进细胞ROS的积累,并伴有线粒体功能障碍,最终导致细胞发生铁死亡[38]。
此外,与其他系统的恶性肿瘤不同的是,血液系统恶性肿瘤患者由于正常红细胞生成障碍及化疗而需要反复输血治疗,导致体内铁负荷加重。而过量的铁和ROS催化产生会通过烟NADPH氧化酶(NOX)和GSH消耗促进造血干细胞的恶性转化[39],且在骨髓增生异常综合征中,ROS诱导的DNA损伤可能增加患者进展为白血病的风险[40]。
上述研究结果表明,过量的铁通过ROS途径促进细胞铁死亡的发生,铁蛋白代谢相关途径介导的细胞内铁离子水平变化也与铁死亡密切相关,调节铁的代谢稳态可影响AML细胞对铁死亡的敏感性,而白血病的疾病进展也会增加患者体内铁的蓄积。
3 总结与展望
铁死亡是由Erastin、RSL3等小分子诱导的一种细胞死亡模式,由GPX4、GSH代谢、铁代谢等多通路调控,其发生发展伴随着ROS的堆积,导致细胞膜发生脂质过氧化,其具体发生机制还需进一步研究。细胞死亡模式的研究仍是攻克治疗肿瘤难题的重要环节之一,近年来铁死亡在肿瘤领域的研究中较为热门,许多研究表明,通过调节铁死亡诱导因子水平、细胞内ROS产生和消亡的平衡以及调控铁代谢稳态能增加部分白血病及淋巴瘤细胞对铁死亡的敏感性,从而达到杀死肿瘤细胞的作用,也发现了多种化合物与肿瘤细胞发生铁死亡密切相关,且铁死亡诱导因子水平与疾病的预后相关。而血液系统肿瘤疾病进展及治疗带来的变化也会对铁死亡过程产生影响。但现有的铁死亡在血液肿瘤中的研究仍处于起步阶段,尚不成熟,需要进一步的体外实验验证铁死亡对血液肿瘤细胞的作用及机制,期望将来能有更多相关研究,为白血病及淋巴瘤的治疗方案提供新的思路。
