基于简单重复序列标记的药用梅品种的身份证构建
2020-12-08欧金梅李肖莉方成武
王 瑞,欧金梅,2,李 昕,李肖莉,张 伟,2,方成武,3
(1.安徽中医药大学,安徽 合肥 230012;2.中药饮片制造新技术安徽省重点实验室,安徽 合肥 230012;3.中药研究与开发安徽省重点实验室,安徽 合肥 230012)
中药梅花与乌梅为蔷薇科梅PrunusmumeSieb. et Zucc.的干燥未成熟果实或未开放花蕾。乌梅药用敛肺涩肠、生津安蛔[1]79,食用开胃生津;梅花疏肝和中、化痰散结[1]265。梅作为药用、食用兼具观赏价值为一体的优良树种,受到广泛关注。
梅起源于中国[2],栽培历史悠久,资源丰富,在全国大部分地区均有分布,其中四川、福建、云南、安徽等省份为药用梅的主产区[3]。梅的栽培品种众多,多以用途进行品种培育和分类,常分为花梅、果梅、药用梅等品种[3-4]。在药用梅的主产区,盲目引种现象比较严重,药用梅与果梅品种混杂,导致药用梅种质混乱。品种在遗传物质上的变化,导致植物在药材性状、产量和质量上均会出现明显的差异[5-6],从而造成中药疗效的不稳定。因此,需建立能将不同种质有效区分的方法,理清种源,为药用梅的品种选育提供科学依据。
DNA身份证是以DNA分子标记技术为基础,通过电泳图谱将种质或品种进行彼此区分的数字串码,目前已广泛应用于物种起源、种质鉴定和遗传图谱构建等领域[7-11]。简单重复序列(simple sequence repeats,SSR)、限制性内切酶片段长度多态性(restriction fragment length polymorphism,RFLP)、扩增片段 长度多态性(amplified fragment length polymorphism, AFLP)、单核苷酸多态性(single nucleotide polymorphism, SNP)等已应用于梅的分类研究中[12-14],其中SSR标记因其共显性遗传、多等位基因性质,已成为品种鉴定和DNA指纹图谱构建的重要途径[15-17]。本研究利用SSR标记技术,采用16对SSR引物建立药用梅30个农家种的DNA分子身份证,通过计算不同品种间遗传相似系数对农家种间关系进行分类,为药用梅的品种鉴定和选育、中药质量控制方法的完善提供依据。
1 材料与方法
1.1 植物 研究材料采于2018年4-7月,收集30个药用梅农家种(见表1),每个农家种采集10个体,共计300份样本,采集嫩叶用硅胶干燥后储存在-70 ℃超低温冰箱备用。样品经鉴定为梅P.mume,凭证标本保存在安徽中医药大学中药资源中心。
1.2 方法
1.2.1 DNA提取 30份样品叶片采用改良十六烷基三甲基溴化铵(hexadecyl trimethyl ammonium bromide, CTAB)法[18]提取总DNA。经1%琼脂糖凝胶电泳检测质量和浓度后,调整浓度为50 ng/μL,保存于-20 ℃冰箱待用。
1.2.2 SSR引物筛选和PCR扩增 本研究从前人研究[13,19-20]中选取了180对SSR引物供筛选,最终选取16对多态性高、条带清晰且重复性好的SSR引物,见表2。
运用Fragment AnalyzerTM96系统以及16对SSR引物对30份样品进行扩增。PCR反应体系:模板DNA 1 μL,正反引物(10 mmol/L)各0.5 μL,MIX酶5 μL,补加双蒸水3 μL,使总体系为10 μL。反应条件:95 ℃预变性4 min,95 ℃变性30 s,按表2温度退火30 s,72 ℃延伸1 min,35次循环,72 ℃延伸6 min。
1.2.3 数据收集和分析 应用PROSizeTM2.0软件通过毛细管电泳的图谱选择清晰的多态性DNA条带进行评分。通常每个个体获得一个或两个片段用于一个SSR标记。尽管所选择的样品都是二倍体,但是对于某些个体,一些标记物扩增了两个以上的片段。基于几个标准仅收集了两个片段:选择较高的峰值,较高的DNA产物的浓度,以及在其他个体中发生的片段频率。使用PowerMarker[21]版本计算等位基因数(effective number of allele,NA)、

表1 30份药用梅植物材料
最小等位基因频率(minor allele frequency,MAF)、观察杂合度(observed heterozygosity, HO)和多态信息含量(polymorphism information content,PIC)。使用Nei的遗传距离构建树状图,并由MEGA7[22]观察。为评估这些标记的指纹识别能力,计算同一性的概率(probability of identity, PI),使用Genalex 6.5对每个标记及其组合进行测试。PI是两个具有相同基因型的随机个体的平均概率。PI的计算公式:PI=2(∑Pi2)2-∑Pi4。其中Pi表示一个位点上第i个等位基因的频率。对于多位点组合,假设所有位点独立分离PI被计算为单个位点PI的乘积。此外,还计算了同胞群体中PI值(probability of identity sibs, PIsibs),从一个种群中找到两个相同个体的总概率,并计算具有相同基因型的突变体。PIsibs计算公式:PIsibs=0.25+(0.5∑Pi2)+[0.5(∑Pi2)2]-(0.25∑Pi4)。其中Pi表示一个位点上第i个等位基因的频率。

表2 筛选的SSR引物信息
1.2.4 核心SSR标记选择 为获得可靠的核心SSR标记集,采用以下标准[10]:①扩增产物明确,无非特异性扩增;②该标记多态性程度较高,PIC>0.5;③相邻等位基因片段大小差异大于3 bp,以便通过凝胶电泳解析;④PI<0.2,使一些标记结合起来可以提供足够的判别能力。
2 结果
2.1 SSR引物的多态性 使用Fragment AnalyzerTM96以及16对SSR标记鉴定30份药用梅农家品种。随后,计算这些标记的遗传特性,包括NA、MAF、HO和PIC。见表3。从30份药用梅种质DNA中共扩增出114个SSR等位基因。毛细管电泳检测系统检测到的等位基因数量从4个(PMSSR0302,PMSSR0346,PMSSR027)到11个(PMSSR024),每个SSR引物对平均可以扩增出7.12个。每个标记的MAF范围为0.20(PMSSR092)至0.76(AG10),平均值为0.39。HO为0.11(PMSSR0302)至0.95(PMSSR0626),平均值为0.72。PIC值为0.51(AG10)至0.85(PMSSR092),平均值为0.69。本研究筛选的16个SSR引物标记位点均为高度多态性位点,表明这些SSR位点具有丰富的遗传差异性,可以有效构建30份药用梅农家种的身份证。
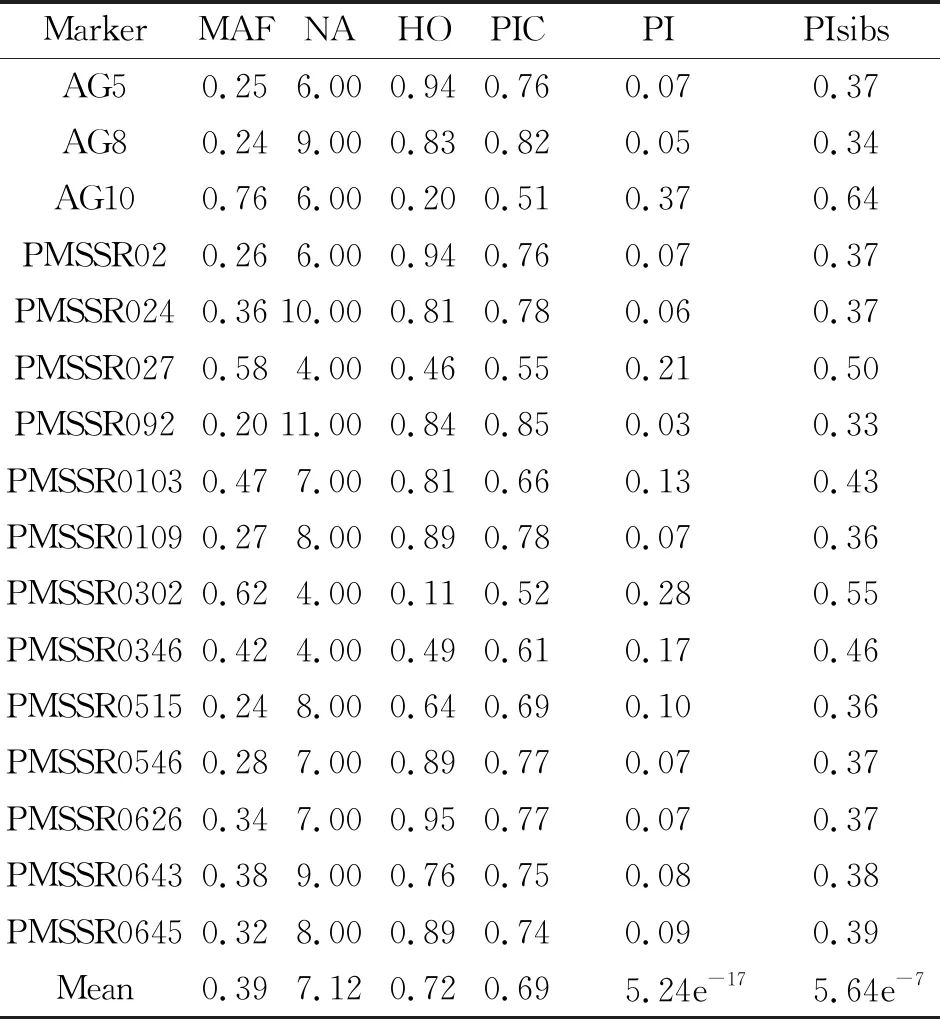
表3 SSR多态性引物的特征
2.2 药用梅DNA身份证的构建 为评价16对标记的指纹识别能力,计算PI和PIsibs两个关键统计量。每一个SSR标记的PI值在0.03(PMSSR092)到0.37(AG10)之间,平均值为0.12。假设所有SSR标记位点独立分离,在所有16个位点上识别两个具有相同基因型的随机个体的概率估计为5.24×10-17。PIsibs从0.33(PMSSR092)到0.55(PMSSR302),平均值为0.41,组合的PIsibs为5.64×10-7。PI和PIsibs值均极低,表明这些SSR标记组合具有良好的指纹识别能力。根据结果,共筛选出4对SSR标记作为核心引物,并提供了明确片段扩增,见表4。因此,将毛细管电泳图谱上的在同一峰值处有带记为1,无带记为0,据此构建30个药用梅农家种DNA身份证,见表5,部分引物毛细管电泳图谱见图1。
2.3 系统发育树的构建 根据Nei遗传相似系数采用非加权算术平均连锁法(unweighted pair group method with arithmetic mean, UPGMA)进行聚类分析 ,如图2所示。在遗传相似系数为0.66时,可将30份药用梅分为3类。第一类由大青梅、达州实生梅、桃梅等7个品种组成,第二类由黑叶青竹梅、绍安土白梅、软蒂青梅和绍安白梅等7个品种组成,第三类由单瓣杏梅、大叶青、江梅、上虞青梅、歙县青梅等16个品种组成。

表4 核心引物扩增片段长度
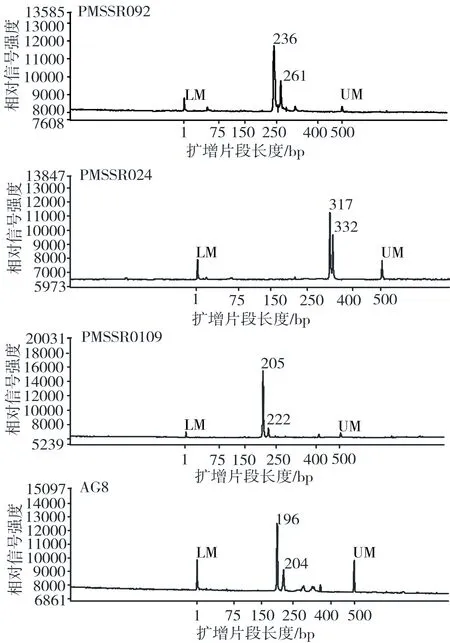
注:最小标记片段(low marker,LM);

表5 部分农家种的DNA身份证
3 讨论
3.1 药用梅DNA的身份证构建 目前,在产地多利用形态学特征对药用梅的种质进行鉴别,而对于跨地区引种和亲缘关系较近的药用梅品种则很难区分。SSR分子标记构建的中药材DNA身份证可以有效区分近缘物种和农家种[23]。本研究采用四川、云南和福建等主要产地的30份药用梅农家种,所构建的30份农家种DNA身份证,可为药用梅的种质数据库的建设奠定基础。未来还将不断扩大种质资源数量和范围,进一步调整和优化SSR引物,最终构建完善药用梅的种质资源DNA指纹图谱库,为药用梅的种质鉴定提供准确、可靠的方法及溯源信息平台。
3.2 SSR引物多态性分析 SSR在植物遗传学和育种中发挥重要作用。然而,SSR的有效性在很大程度上依赖于标记的质量、基因分型数据的准确性和所选择的植物材料。本研究从180对SSR引物中筛选出的16对标记在30份药用梅中具有较好的多态性。16对SSR标记的PI值在0.03(PMSSR092)到0.37(AG10)之间,平均值为0.181,在所有16个位点上识别两个具有相同基因型的随机个体的概率估计为5.24×10-17,PI为0.000 1~0.01[21-22],已足够鉴定大多数自然群体,表明这些SSR标记组合具有良好的指纹识别能力。每个标记平均有7.12个等位基因,平均PIC值为0.69。平均NA和PIC值略高于前人研究结果[19,24],原因可能是位于非转录区的某些SSR序列具有相对较低水平的序列保守性或者更高的突变性,或是因为毛细管电泳具有更高的分辨率,对于样品中高通量片段的分离、等位基因的鉴别具有特别优势。
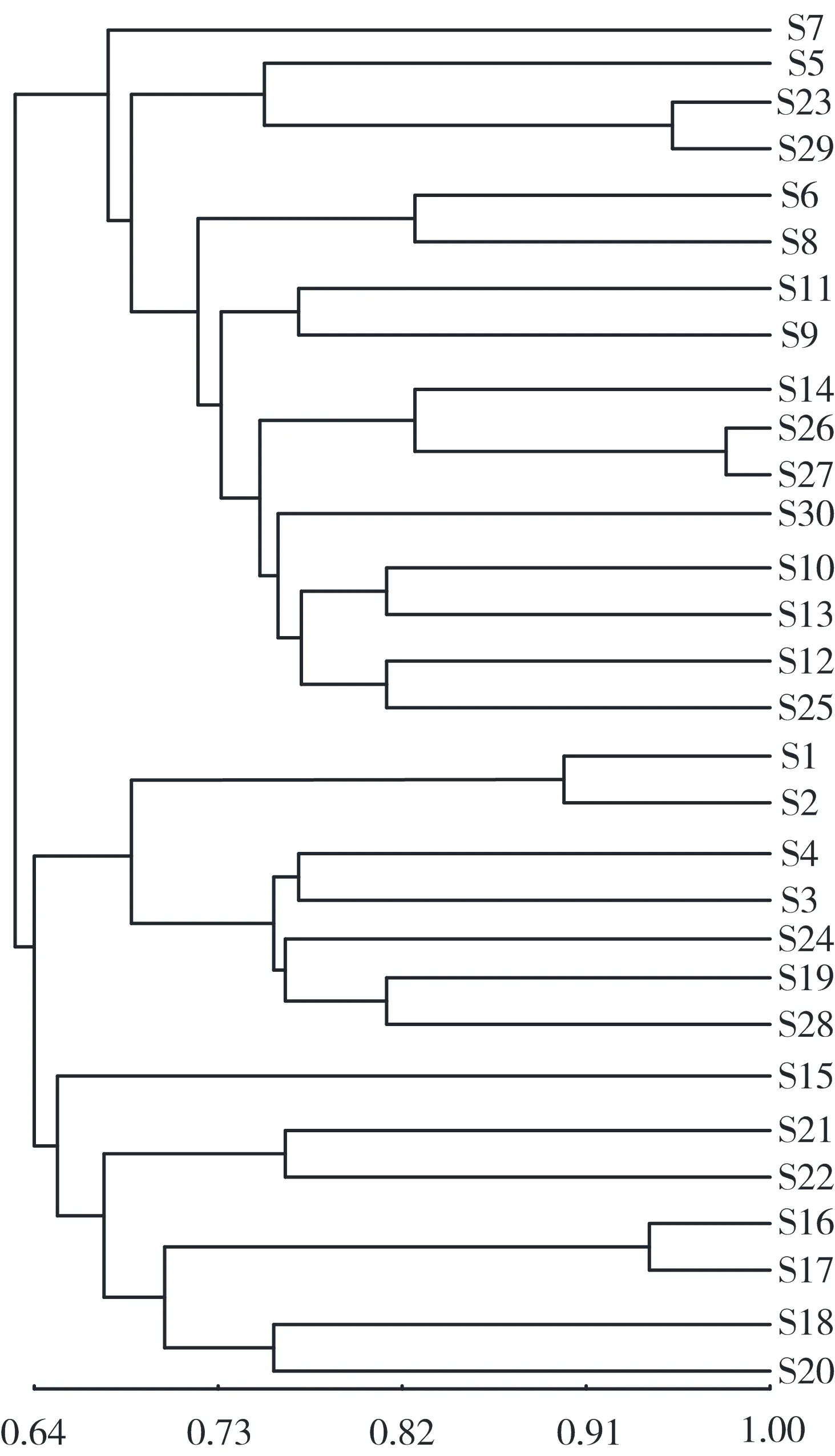
图2 30份药用梅农家种的聚类图
3.3 系统发育树分析 本研究使用基因组SSR对30份药用梅农家种进行遗传多样性研究,其平均多态信息含量较高,表明药用梅的遗传多样性水平较高。基于UPGMA聚类图可以发现,样品可聚为三大支,相同来源的种质之间亲缘关系较为较近,如第三类中云南丽江的26(MSH14)和27(MSH13)样品,表明其遗传背景高度一致。第二类中的1(黑叶青竹梅)、2(绍安土白梅)、3(软蒂青梅)和4(绍安白梅)聚为一类,因其产地相同,推测其具有相似的遗传背景。而12(歙县青梅)和13(临安青梅)与3(上杭青梅)距离较远。其遗传一致性降低,可能与种质差异或引种后种质混乱导致遗传变异有关。目前,SSR标记扩增的序列具体与药用梅的哪个性状相关联尚不清楚。且SSR标记的数字化具有局限性,不能反映一个药用梅品种的全部信息。因此,之后的研究应将SSR标记与植物的表现型相结合,以完善药用梅的品种鉴定技术体系,为药用梅的优良品种保护、溯源管理等提供理论依据。
4 结论
本研究是基于前人研究的180对SSR引物筛选出多态性高、稳定性好的16对SSR引物,并对30个药用梅的农家种进行SSR扩增分析,共检测到114个等位基因,平均NA为7.12个,平均PIC和PI信息指数分别为0.69和5.24×10-17。最终确定引物PMSSR092、PMSSR0109、PMSSR024和AG8为核心引物,4对引物组合可区分供试品种。
