硝化酶的研究进展
2020-12-04赵璐瑶陈振娅霍毅欣
赵璐瑶 陈振娅 霍毅欣,2
(1.北京理工大学生命学院,北京 100081;2.苏州工业园区洛加大先进技术研究院,苏州 215123)
硝基是强钝化基,是炸药、染料、生物医药和抗菌剂中常见的官能团[1-2]。硝基化合物是烃分子中的氢原子被强电子受体-NO2取代而成的化合物,根据脂烃基或芳烃基可分为脂肪族硝基化合物(RNO2)和芳香族硝基化合物(Ar-NO2),如硝基甲烷、硝基苯。根据含硝基的数量,又可分为一元硝基化合物和多元硝基化合物,如对硝基苯(NB)、三硝基甲苯(TNT)[3]。芳香族硝基化合物因其安全性好、感度较低、能量适中,在很多领域[4],尤其在炸药制造中是一类非常重要的原料[5](图1)。

图1 硝基化合物的制备及转化
目前,化学合成法是硝基化合物的主要合成方法[5-7],利用硝酸和硫酸作为硝化试剂可顺利得到多硝基化合物,但同时会产生大量含有机化合物的废酸和废水,造成严重的环境污染[8]。有许多优化的硝化方法[9],例如利用硝酸盐等重氮盐取代酸或者用镧系(Ⅲ)或(Ⅳ)族金属[10]、三氟磺酸[11]等作为催化剂,这些方法虽在一定程度上缓解了环境污染问题,但在成本、可控性和转化效率上仍有不足。生物酶催化高效环保,反应条件温和,耗能少,副产物少,可减少环境污染并降低分离产物所需成本[12],生物酶合成法在生产领域的应用越来越广泛。本文总结了硝基及硝基化合物的功能,当前已有的硝基化合物合成方法以及包括过氧化物酶、细胞色素P450超家族和N-加氧酶等硝化酶的研究进展,展望了利用硝化酶催化芳香族硝基化合物的合成以及通过优化反应条件、酶的定向进化提高酶催化效率、创造新功能酶的发展方向。
1 硝基及硝基化合物
硝基是炸药、染料、生物医药和抗菌剂等常见的官能基团[1-2,13]。硝基化合物是烃分子中的氢原子被-NO2取代而成的化合物,其通式为R-NO2或Ar-NO2,如硝基甲烷、硝基苯等,其中芳香族硝基化合物尤为重要[14]。硝基化合物在生产和生活中具有广泛的应用,Abdallah等[3]证实了芳香族硝基化合物具有降低碳钢的阳极溶解、延缓阴极析氢反应的作用,可用作抑制碳钢腐蚀的混合抑制剂。Ono等[15]利用硝基烯烃与异氰酸酯反应合成不含N的吡咯,Dryzhakov等[16]探究了硝基化合物在催化叠氮化反应中的协同催化作用。另外,硝化反应也是制备氨基化合物的一条重要途径[17],不同的还原剂可以使硝基化合物生成各种不同的中间还原产物[18],且这些中间产物在一定条件下又可以互相转变[19]。
2 化学硝化方法
传统化学合成方法中最常见的是利用硝酸、硫酸等强酸制备硝基化合物[8],但会产生大量对环境有害的废酸和废水,且副产物较多[20]。为解决以上问题,可通过改变催化剂种类、调整酸的使用比例和使用量以及改变反应条件等方法改进硝基化合物的生产过程[21]。有些硝基化合物如邻二硝基苯、对二硝基苯均不能由酸直接硝化,只能由邻硝基苯胺、对硝基苯胺形成的重氮盐与亚硝酸钠反应制得(图2)[22-23]。较为理想的方法是用镧系(Ⅲ)或(Ⅳ)族金属[10]、三氟磺酸[11]等作为催化剂以较高产率催化等当量的硝酸(69%)硝化苯环,此反应副产物中没有废酸且催化剂可重复使用,但也存在催化选择性较差,对设备要求高,产生的废物处理过程繁琐以及安全隐患等问题[24]。为拓展反应条件温和、成本低及产生的副产物较少的生物酶合成法在生产领域的应用,需要探究硝化酶的结构和功能。
3 自然界中的硝化酶
自然界中的许多生物体可以发生硝化反应[25],这些硝化反应依赖于其体内酶的催化[26]。已报道的硝化酶大致可分为过氧化物酶、细胞色素P450家族和N-加氧酶3大类。
3.1 过氧化物酶
过氧化物酶是一类氧化还原酶,主要存在于细菌、真菌、植物和动物中,以过氧化氢或烷基过氧化物作为电子受体,可有效催化一系列底物的氧化[27-29]。微过氧化物酶(Micro-peroxidases,MPs)是细胞色素C在胃蛋白酶和胰蛋白酶作用下水解得到的血红素肽链,结构简单并保留了过氧化物酶的活性。包括含血红素的十一肽(第12-22位氨基酸残基),即微过氧化物酶11(MP11)[30]、含血红素的十肽(第13-22位氨基酸残基),即微过氧化物酶10(MP10)[31]、含血红素的九肽(第14-22位氨基酸残基),即微过氧化物酶9(MP9)[32]和含血红素的八肽(第14-21位氨基酸残基),即微过氧化物酶8(MP8)[33]。其中,MP8的研究最为广泛。Aron等[34]从马心细胞色素C中降解MP8。Marques等[35]对MP8的水化合物有机溶液反应进行了全面的研究,发现MP8具有过氧化物酶活性,能够氧化N-单取代羟胺。Ricoux等[36]利用MP8催化苯酚在NO2-/H2O2/MP8体系中合成邻硝基苯酚和对硝基苯酚。MP8保留了细胞色素C第14-21位氨基酸残基,其中C14和C17通过硫醚键与血红素桥联,铁离子占据了His18上咪唑基团的第六位点,水分子占据了轴向配位的位点,这使得MP8可与底物苯酚的烷基结合并形成硝基[37]。Buddea等[38]报道了大豆过氧化物酶(Soybean peroxidase,SBP)可催化无机化合物的氧化且可在过氧化氢和亚硝酸钠的存在下催化多种酚类的硝化。SBP具有很高的热稳定性和酸碱耐受性,这使其在工业和环境领域中具有广泛的应用价值[39]。Welinder等[40]得到了非糖基化的重组SBP(rSBP),通过比较氨基酸序列观察到rSBP晶体结构中胱氨酸与C11-C91、C44-C49、C97-C299和C176-C208氨基酸序列桥联,这与辣根过氧化物酶C(HRPC)及其他Ⅲ型过氧化物酶的保守序列同源。通过分析酶的晶体结构发现酚类底物的结合位点上的TRIS缓冲分子以及高度暴露的血红素边缘,以及血红素上的乙烯基与蛋氨酸的硫原子相连是该酶催化功能的结构基础。但至今仍未发现高稳定性酶的结构基础。
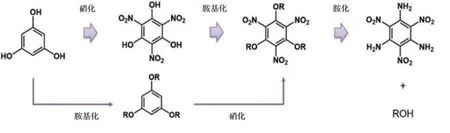
图2 对、邻二硝基苯的合成[22-23]
3.2 细胞色素P450家族
催化有机化合物氧化的P450s中也有催化硝化反应的酶,P450蛋白种类的多样性及其底物的重叠性使P450酶系可以催化多种反应[41]。链霉菌可生产一种叫做萨克多敏E酶(Thaxtomin E,TxtE)的P450酶,该酶可催化芳烃的硝化[42]。由于独特的化学性质,TxtE可能成为设计新型芳香硝基化合物催化剂的理想平台。为了更好地了解野生型酶的结构和功能,Yu等[43]利用Auto Dock证实了三种与色氨酸连接的构造。Dodani等[44]通过光谱和酶-底物拟合研究,确定野生型TxtE可催化L-色氨酸结构类似物,而这些底物需要有被修饰的吲哚侧链和氨基酸基团才能被硝化。
3.3 非血红素二铁N-加氧酶
催化硝化反应的N-加氧酶在自然界中较为稀有,其中研究较深入的是来自土壤细菌Streptomyces thioluteus的对氨基苯基-N-加氧酶(p-aminobenzoate N-oxygenase,AurF),该酶是一种非血红素二铁加氧酶,催化将对氨基苯甲酸(Ar-NH2,Ar=4-羧基苯)经由对羟氨基苯甲酸(Ar-NHOH)、对亚硝基苯甲酸(Ar-NO)转化为对硝基苯甲酸(Ar-NO2)[45]。Choi等[46]分析了AurF的电子传递机制,证实了AurF是由两个铁离子而非锰离子传递电子,并实现了AurF的体外酶促反应(图3)[47]。与AurF同源的另一非血红素二铁酶(α-N-dichloroacetyl-p-aminophenylserinol N-oxygenase,CmlI)也具有催化氨基氧化的作用[48-49]。Lu等[50]首次报道了N-加氧酶CmlI的生化特性以及在氯霉素(Chloramphenicol,CAM)生物合成中的作用。CAM生物合成的最后一步是CmlI将D-苏式-1-(4-氨基苯基)-2-二氯乙酰氨基-1,3-丙二醇(NH2-CAM)的芳胺功能团氧化为硝基。生物信息学分析结构表明,CmlI与AurF中34%的氨基酸序列相同且均含有双铁氧合酶中特有的EX28e37DEXXH基序。Knoot等[51]在研究中报道了AurF第197-201的氨基酸序列处有一段非α-螺旋结构,而CmlI在该处仍为α-螺旋。这些结构上的特征为进一步利用蛋白质工程工具进行此类加氧酶和生物催化剂的机理和结构研究打开了大门。此外,氨基吡咯菌素-N-加氧酶(Monodechloroaminopyrrolnitrin synthase,PrnD)也是一种非血红素铁酶,可催化氨基吡咯尼林转化为吡咯尼林[52]。作为Rieske蛋白,PrnD的2Fe-2S中心和单核铁位点在进化上是保守的,与该酶的2Fe-2S中心配位的C69、H71、C88和H91以及与非血红素铁表面配位的3个氨基酸残基H186、H191和D323可避免线性序列的形成并保证酶的催化活性[53]。

图3 AurF结构分析[46]
4 天然酶的定向改造
天然酶因其本身存在缺陷而难以满足人们的需求。在实际生产中,为得到所需特性的酶,需要分析酶活性区域的结构,改变酶的活性口袋进而提高其催化活性并拓宽底物范围。表1中总结了已报道的硝化酶的改造位点及效果。
4.1 TxtE酶的结构分析与改造
为验证TxtE酶的结构和功能的关系,Yu等[43]设计了5个单残基突变实验验证TxtE与L-色氨酸结合的分子对接模型,将R59、N293、T296、E394突变为亮氨酸,Y89突变为苯丙氨酸后,发现这几个氨基酸对L-色氨酸具有极性作用。为识别底物与突变体的结合情况,在含野生型TxtE的反应体系中加入L-色氨酸后,观察血红素紫外-可见光谱的变化,发现390 nm处的吸收增加,420 nm处的吸收减少,而突变体TxtE-R59L、TxtE-T296L和TxtEE394L在添加L-色氨酸后未见移位,突变体TxtEY89F和TxtE-N293L仍能与L-色氨酸结合,但结合能力明显下降,说明R59、T296和E394是L-色氨酸结合所必需的氨基酸。TxtE-Y89F和TxtE-N293L的解离常数(Kd)分别为120.63 ± 4.30 μmol/L和1.70±0.28 μmol/L,而 野 生 型TxtE的Kd值 为43.59±2.26 μmol/L。因此,N293也可能是结合L-色氨酸的关键残基。为提高酶的催化活性,Zuo等[54]构建了15个TxtE融合体。将TxtE的JK环与P450BM3的JK环进行交换,调整TxtE与BM3R之间的连接肽长度(图4)并获得多个融合体,其中TB14(连接肽长度为14个氨基酸)催化活性明显高于TxtE,且底物范围广,可催化4-F-5-NO2-L-Trp和4-Me-7-NO2-L-Trp的合成。TB14在生物医学和生物应用领域中具有广泛的应用前景,是开发硝化生物催化剂、通过蛋白质工程合成结构多样的芳香族硝基化合物的理想材料。

表1 硝化酶的改造及效果

图4 含有长度可变的连接肽或交换环(黄色)的TxtEBM3R嵌合体结构示意图[54]
Saroay等[55]进一步研究并发现TxtE对吲哚环取代等底物修饰具有耐受性,为提高底物的转化率并扩大底物范围,他们对22个位点饱和文库进行了天然底物筛选,得到了R59X突变体,该突变体催化色氨酸生成4-硝基色氨酸的转化率比野生型提高了3.5倍。Gober等[56]通过在H176位点进行饱和突变以探索氨基酸残基的替换对区域选择性的影响,多数突变体的活性显著降低甚至完全失活,但也发现少数突变体的催化活性提高或者催化位点改变。如将H176替换为为天冬酰胺,甘氨酸、丝氨酸、半胱氨酸或蛋氨酸后,酶可以催化4-和5-硝基色氨酸的合成,替换为苯丙氨酸、酪氨酸则只在色氨酸的C5位产生硝化作用。硝基色氨酸及其类似物是合成生物活性和生物技术相关的化学物质、材料和蛋白质的基础材料,对TxtE底物范围和区域特异性的分子决定因素的结构和机制的研究,可扩展对酶的基本理解,有利于指导进一步的工程研究。
4.2 非血红素二铁N-加氧酶的结构分析与改造
Lee等[53]为探讨荧光假单胞菌Pf-5中氨基吡咯硝基加氧酶(PrnD)底物特异性的决定因素。采用分子模拟和突变分析(定点诱变和饱和诱变)方法推测出17个可能与底物接触的氨基酸残基,其中第312位和277位氨基酸残基可以单独或共同调节酶底物的特异性。通过用丙氨酸或缬氨酸取代这些氨基酸来探讨它们在底物特异性中的作用,测定不同突变体对底物Aprn的催化活性并与野生型酶对底物的催化活性进行比较,发现多数突变体活性显著下降,该结果证明这些氨基酸对PrnD的底物特异性或催化活性的重要性。此外,该研究还发现F312A和L277A突变体催化底物Aprn的转化效率分别提高了4.89倍和3.28倍,当这两个突变组合在一起时,F312A和L277A突变体表现出累积效应,并对底物Aprn的催化活性进一步提高。Zocher等[57]通过模拟AurF与底物pABA复合物的模型分析了AurF的保守序列,改变AurF活性位点的氨基酸,并分别测定了突变体及野生型的体内酶活。将R96突变为丙氨酸时,AurF对pABA的催化活性完全消失,说明R96是决定酶催化功能的重要氨基酸(图5)。将T100替换成亮氨酸,酶活性急剧下降,说明T100对酶的活性具有重要作用。相反,将T100替换为丙氨酸后酶活性可提高3倍,F264替换为丙氨酸酶活性几乎无变化,这可能由于丙氨酸与pABA间仍存在联系。L202替换为苯丙氨酸酶活性提高了3-4倍,这可能由于与苯丙胺酸sp2 Cδ原子共同释放的L202 sp3 Cδ原子使酶与底物之间的联系更加紧密。这些突变体的活性验证了AurF的底物结合模型(图5),基于酶结构可利用AurF的定向改造,使其成为一种功能更加完善的生物催化剂。另外,Choi等[46]验证了AurF可催化氨基芳烃的连续氧化,该酶由主要结构为α螺旋的单体A和单体B组成的同源二聚体,并结合生化和结构分析证明AurF是一种非血红素二铁加氧酶。蛋白残基 E101、E136、H139、E196、H223、E227和H230通过配位键与两个铁离子连接形成活性中心。此外,还报道了氧化状态下的晶体结构及与产物pNBA的共晶结构。这为进一步研究AurF的催化机制奠定了坚实的基础。
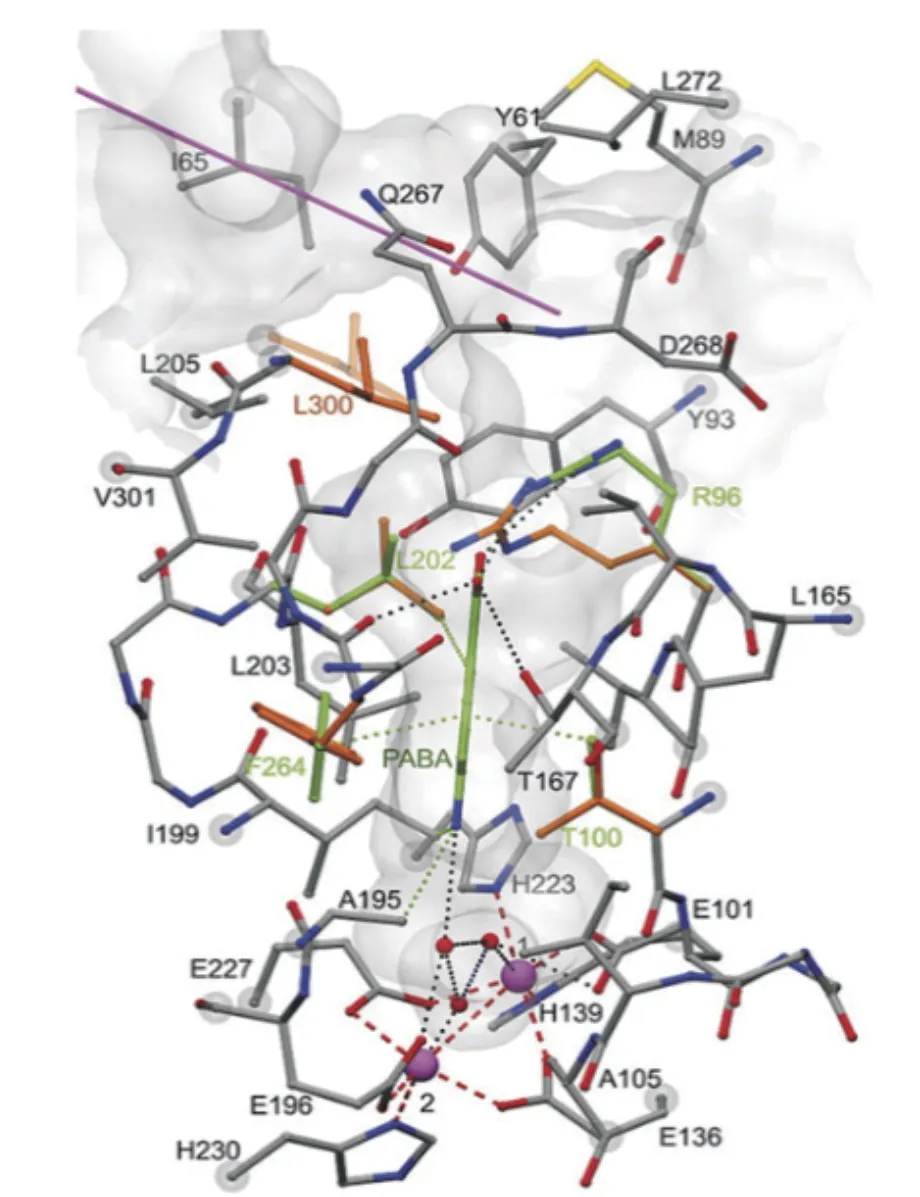
图5 使用已建模的底物pABA(绿色)观察AurF亚基活性中心的立体结构[57]
5 硝化酶的应用实例
抗生素吡咯尼特林及多拉菌素的生物合成是酶催化芳胺氧化的最典型的实例[58]。大肠杆菌可表达与吡咯尼林生产相关的4种功能不同的酶PrnA、PrnB、PrnC和PrnD[59]。Pée等[60]提出了吡咯尼林的生物合成途径,首先PrnA催化色氨酸生成7-氯色氨酸,然后吲哚环重排形成苯基吡咯环并在PrnB的作用下脱羧生成单氯氨吡咯尼林,经PrnC二次氯化生成氨基吡咯尼林,最后PrnD催化氨基吡咯尼林(Aprn)的氨基氧化成硝基,形成吡咯尼林。此外,He等[61]验证了聚酮合成酶(PKS)以对硝基苯甲酸(pNBA)为反应前体这一特有功能,利用AurF通过N-氧化作用催化对氨基苯甲酸得到合成多拉菌素的前体pNBA,并进一步在PKS的作用下合成终产物多拉菌素。
6 总结与展望
本文首先综述了硝基基团以及硝基化合物的重要作用,总结了硝基化合物(主要为芳香族硝基化合物)合成方法的种类以及合成工艺的缺陷。另外,提供了生物酶合成化工产品的新思路,总结了硝化酶(p450超家族、过氧化物酶以及N-加氧酶)的研究进展,归纳了通过改变酶的活性口袋进而提高酶的催化活性并拓宽底物范围的研究方法,为解决化工生产中面临生产效率低、安全性低、转化效率低以及高成本、高污染和高设备要求等关键问题提供了思路。尽管对硝化酶研究大多还停留在酶的结构分析、催化机理和酶的活性及稳定性的优化上,在实际应用中的例子十分罕见,但硝化酶在生产领域中将会带来十分重要的经济价值以及广阔的发展空间,可通过以下几种方法来进一步扩展研究:(1)通过计算机模拟,分析酶和底物复合物的晶体结构,明确碳硝化酶的底物催化识别机制,对活性口袋关键氨基酸进行突变,实现氮硝化酶的定向进化;(2)将催化硝化反应的酶与其他有利功能的酶的表达基因进行重组,从而表达出全新催化功能的融合酶;(3)进一步挖掘自然界中的微生物酶源,从而获得更多具有催化芳香族硝化的酶。其中,前两种方法都是从结构上对酶进行改造,从而创造出一种符合人们期望的具有新的催化活性的酶。硝化酶的研究和改造可为进一步扩大酶的生产,为生物法取代传统化学方法制备炸药、医药、染料等奠定基础。
