S100A8/A9在头颈部肿瘤发生发展中的作用及机制
2020-12-02冀慎英张湘豫邹先琼
冀慎英,张湘豫,邹先琼
(1.桂林医学院附属口腔医院口腔医学研究所,广西 桂林 541004; 2.桂林医学院基础医学院,广西 桂林 541100)
头颈部肿瘤是来源于口腔、鼻腔、咽及喉等部位的恶性肿瘤的总称,常见类型包括口腔鳞状细胞癌(oral squamous cell carcinoma,OSCC)、口咽鳞状细胞癌(oropharyngeal squamous cell carcinoma,OPSCC)、食管鳞状细胞癌(esophageal squamous cell carcinoma,ESCC)、鼻咽鳞状细胞癌(nasopharyngeal squamous cell carcinoma,NSCC)、喉鳞状细胞癌(laryngeal squamous cell carcinoma LSCC)、甲状腺癌(thyroid cancer,TC)等[1-2]。OSCC是最常见的头颈部鳞状细胞癌(head and neck squamous cell carcinoma,HNSCC),常伴有口腔癌前病变,如口腔白斑等[3]。随着研究的深入发现,S100A8/A9的异常表达与头颈部肿瘤的发生发展密切相关[1]。
1 S100A8/A9的结构与功能
S100A8和S100A9均属于S100蛋白家族,分子量分别为10 800和13 200[4],其分子结构均由两个不同的螺旋-环-螺旋配基组成,两侧为疏水区,中央为铰链区,铰链区与其他靶蛋白结合[4]。S100A8及S100A9均为酸性钙离子结合蛋白,两者以钙离子依赖性方式形成异源二聚体S100A8/A9(即钙防卫蛋白)[5]。S100A8/A9能诱导中性粒细胞趋化和黏附,促进白细胞花生四烯酸的转运和代谢,调节吞噬细胞迁移和中性粒细胞还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶活化过程中微管蛋白依赖性细胞骨架的代谢,具有促炎、抗菌、抗氧化和诱导凋亡等功能[5]。
S100A8/A9以组织特异性方式表达,在多种肿瘤的发生发展过程中发挥重要作用[1]。S100A8/A9在皮肤癌、乳腺癌、前列腺癌、卵巢癌、胰腺癌、结直肠癌、膀胱癌、胃癌[6]以及部分TC组织[1]中高表达,具有促进肿瘤发生发展的作用[1]。S100A8/A9在OSCC、OPSCC、ESCC、NSCC及LSCC等头颈部肿瘤组织中低表达,一般认为发挥了抗肿瘤作用[1]。S100A8/A9在不同肿瘤组织中的作用可能取决于其在细胞内外的水平和位置[5]:其在细胞外高水平时能诱导细胞凋亡;细胞内高水平时可调节上皮细胞间质化与间质细胞上皮化之间的信号级联,诱导癌细胞侵袭能力降低[5]。
2 S100A8/A9在头颈部肿瘤发生发展中的作用
2.1S100A8/A9与OSCC和OPSCC S100A8/A9表达失调是口腔和口咽肿瘤发生的一个特征[1]。在OSCC、OPSCC细胞中S100A8/A9的表达水平较口腔和口咽非肿瘤性复层鳞状上皮显著降低[1]。S100A8/A9的表达在高分化、中等分化、低分化以及非角质化基底细胞样癌HNSCC中逐渐降低,提示S100A8/A9的表达水平在HNSCC的发生发展中逐渐下调[7]。一般认为OSCC中S100A8/A9表达的下调与肿瘤分化不良[1]以及DNA甲基化增加[8]有关。
S100A8/A9可能调节OSCC细胞的恶性特征[1]。在高分化的人OSCC细胞系中,采用短发夹RNA沉默内源性S100A8/A9后,细胞基质金属蛋白酶(matrix metalloproteinase,MMP)-2的活性及细胞迁移和侵袭能力增强[9];相反,当S100A8/A9在KB细胞(S100A8/A9阴性表达的细胞)过表达后,MMP-2的表达、细胞迁移及侵袭能力减弱[9],表明S100A8/A9能够负性调控MMP-2蛋白的产生和相关酶的活性。另一方面,在MC38结肠癌细胞和LLC肺癌细胞中,S100A8/A9的下调显著降低了MMP-2和MMP-9在癌细胞中的表达,进一步导致癌细胞迁移和侵袭能力降低[10],说明基质来源和肿瘤来源的S100A8/A9对于调节MMP-2和MMP-9的表达有功能差异。用单核/巨噬细胞条件培养基诱导MC38和LLC细胞表达S100A8/A9,与未表达S100A8/A9的MC38和LLC癌细胞相比,细胞内钙离子水平降低[9]。当诱导的S100A8/A9被敲除时,癌细胞中钙离子水平降低的现象被消除[9],提示S100A8/A9可通过调节癌细胞内钙离子的水平调节MMP-2、MMP-9的表达[9]。使用小鼠异种移植肿瘤模型发现,KB细胞和KB-EGFP细胞形成的肿瘤明显大于KB-S100A8/A9细胞[8],说明S100A8/A9与控制细胞发育和分化、细胞间信号和相互作用以及细胞形态的基因网络相互作用,下调了与侵袭和肿瘤发生相关的基因的表达[8]。
研究发现,S100A8/A9在OSCC中表达的下调与G2/M期细胞周期阻滞密切相关[11]。典型的G2/M细胞周期检查点信号通路受细胞周期检测点激酶1(checkpoint kinase 1,Chk1)的控制[11]。细胞周期中的细胞分裂周期蛋白25同源蛋白C(cell division cyclin 25 homolog C,Cdc25C)是激活细胞周期蛋白B1(cyclin B1)/细胞周期蛋白依赖激酶1复合物和进入有丝分裂所必需的[11]。S100A8/A9的表达增加了Chk1(Ser345)的磷酸化和蛋白磷酸酶2A的活性,p-Chk1(Ser345)使Cdc25C在Ser216处磷酸化后与分子伴侣14-3-3β结合,形成无活性的p-Cdc25C/14-3-3b复合物,导致Cdc25C失活并在胞质中积聚[11]。另一方面,p-Cdc25c(Thr48)可使p-Cdc2(Thr14/Tyr15)去磷酸化成为有活性的Cdc2,Cdc2与cyclin B1结合成为cyclin B1/Cdc2复合物,从而进行有丝分裂[11]。但Cdc25C的Thr48和Ser216残基不能同时磷酸化[11]。当蛋白磷酸酶2A活性增强后可使p-Cdc25c(Thr48)去磷酸化,导致p-Cdc2(Thr14/Tyr15)积聚,cyclin B1表达下调,细胞周期在G2/M检查点停止,导致细胞周期阻滞[11](图1A)。
表皮生长因子受体(epidermal growth factor receptor,EGFR)在OSCC、OPSCC中高表达[7]。EGFR可被胱天蛋白酶(caspase)1、3和7水解和翻译后修饰,故细胞表面EGFR的变异可能反映caspases-1、caspases-3和caspases-7的裂解[7]。S100A8/A9在头颈部肿瘤细胞中过表达时,caspase-1、caspases-3和caspases-7的转录水平和活性提高,导致EGFR水解作用增强,EGFR水平降低,进而影响EGFR依赖的下游信号转导[7]。在TR146细胞沉默S100A8/A9后,细胞caspase-3和caspase-7的活性显著降低,而EGFR水平升高,这说明S100A8/A9的表达与EGFR蛋白呈负相关[7](图1B)。该研究也表明S100A8/A9相关的EGFR下调可能有助于S100A8/A9高表达的HNSCC患者总生存率的提高[7]。
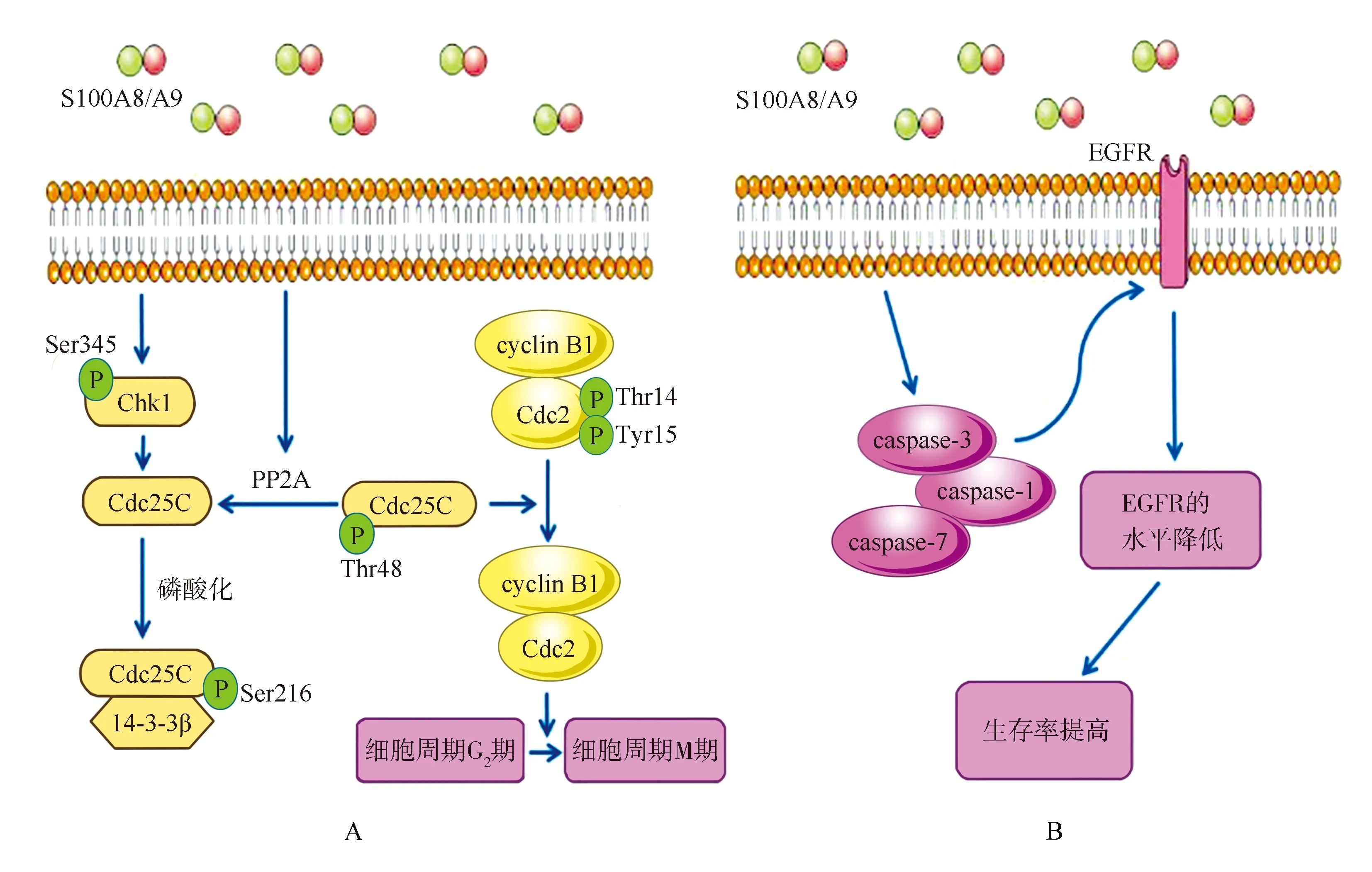
OSCC:口腔鳞状细胞癌;p-Chk1(Ser345):在丝氨酸345处磷酸化的周期检测点激酶1;Cdc25C:细胞分裂周期蛋白25同源蛋白C; p-Chk1(Ser216):在丝氨酸216处磷酸化的周期检测点激酶1;14-3-3b:分子伴侣;PP2A:蛋白磷酸酶2A;p-Chk1(Thr48):在苏氨酸48处磷酸化的周期检测点激酶1;cyclin B1:细胞周期蛋白B1;p-Cdc2(Thr14/Tyr15):在苏氨酸48处或酪氨酸15处磷酸化的细胞周期蛋白依赖激酶1; EGFR:表皮生长因子受体;caspase:胱天蛋白酶
在人乳头瘤病毒(human papillomavirus,HPV)阴性、S100A8/A9高表达的OSCC、OPSCC细胞中,S100A8/A9减弱了癌细胞的恶性表型,起到了抑癌作用[7]。S100A8/A9可能通过调节酪蛋白激酶Ⅱ介导的体外HPV E7蛋白的磷酸化而抑制病毒的致癌活性[12]。HPV诱导的S100A8/A9的间接下调可能是一种额外的致瘤机制[7]。
2.2S100A8/A9与ESCC 在ESCC中S100A8/A9表达下调,且其表达水平与ESCC的分化程度呈负相关[13]。低分化ESCC组织细胞与中度和分化良好的ESCC组织细胞相比,S100A8/A9的表达显著下调,甚至不表达[13],这可能与S100A8/A9在食管鳞状上皮细胞中能阻滞细胞周期进程,进而抑制细胞生长有关[13]。
4-硝基喹啉-1-氧化物(4-nitroquinoline-1-oxide,4-NQO)是一种前体致癌剂,在舌部黏膜中含量较高,故4-NQO也可作为舌癌的标志物之一[14]。环加氧酶2(cyclooxygenase-2,COX-2)是合成前列腺素的关键酶,也是核因子κB(nuclear factor-kappaB,NF-κB)的下游靶点[14]。经4-NQO处理的缺锌性小鼠的ESCC中S100A8的免疫反应显著增强[14]。当4-NQO局部诱导缺锌COX-2-/-小鼠发生食管癌时,S100A8/A9在近端胃癌/舌癌中的表达上调[14]。免疫组织化学显示,S100A8/A9、晚期糖基化终末产物受体(receptor for advanced glycosylation end product,RAGE)、NF-κB、p65和cyclin D1在缺锌性小鼠近端胃或舌的癌前病变和癌中共表达,提示RAGE-S100A8/A9炎症信号通路在近端胃/舌的癌前病变和癌变中被激活[14]。因此,缺锌可能调节S100A8/A9的表达,并调节S100A8/A9与RAGE的相互作用和S100A8/A9与下游NF-κB/COX-2信号通路间的联系,从而促进食管细胞增殖和癌变[14](图2A)。通过特殊饮食建立缺锌性增生和补充锌的大鼠食管组织模型发现,S100A8/A9在缺锌性增生食管中的表达与补锌组织相比显著增强,表明补锌能恢复S100A8/A9基因在体内的表达和食管的生理表型[15]。缺锌性小鼠ESCC的NF-κB通路还受微RNA(microRNA,miRNA)的调控[16]。miR-31是最常见的肿瘤相关失调的miRNA之一,在ESCC、舌鳞状细胞癌等HNSCC组织中高表达[17]。缺锌性小鼠ESCC诱导的癌基因miR-31的过度表达能够下调丝氨酸/苏氨酸激酶40(Stk40),进而通过S100A8/A9激活NF-κB和RAGE,促进炎症发生和细胞增殖,产生食管癌前表型(图2B)[17]。以上研究表明,锌缺乏激活了S100A8/A9-RAGE/NF-κB和miR-31/Stk40/NF-κB信号通路,促进细胞增殖,抑制细胞凋亡,从而促进食管癌的发生发展[17]。
2.3S100A8/A9与NSCC及LSCC 在NSCC组织中,S100A8/A9的表达水平显著低于癌旁正常组织[18]。S100A8/A9在鼻咽恶性肿瘤的上皮中表达不明显,但在非肿瘤性上皮的浅层表达[18]。S100A9在癌细胞和正常咽上皮细胞中的免疫反应性均为阴性,但在所有NSCC的间质炎症细胞中均有免疫反应性[19]。在更晚期NSCC区域淋巴结转移增加中,S100A9的表达在炎症肿瘤微环境中上调,但在癌细胞未上调[19]。S100A8/A9通过剂量效应影响鼻咽癌细胞的增殖与凋亡[20],低水平S100A8/A9(S100A8/A9<10 mg/L)能够促进鼻咽癌细胞株CNE1及CNE2的增殖,而S100A8/A9水平超过30 mg/L则会抑制细胞生长[21]。
在LSCC组织中,S100A9的表达水平显著低于癌旁正常组织[22]。S100A8的3′非翻译区携带一个直接与miR-24结合的特异位点,在LSCC的Hep2细胞中miR-24能在翻译水平直接靶向S100A8,显著诱导Hep2细胞的形态学改变,并在阻断S100A8蛋白后显著抑制Hep2细胞的增殖和侵袭[23];另一方面,circMAN2B2可通过抑制miR-1205促进S100A8的表达,进而促进细胞的增殖、侵袭和迁移[24]。
2.4S100A8/A9与TC TC是来源于内分泌器官的最常见的恶性肿瘤之一,滤泡状癌和乳头状癌是其最常见的两种类型,且均不表达S100A9[25]。S100A9在TC中的阳性率普遍较低,但未分化癌除外[25]。在未分化癌中,S100A9呈较高表达[25]。一般认为,升高的S100A9蛋白可能会导致TC的去分化。
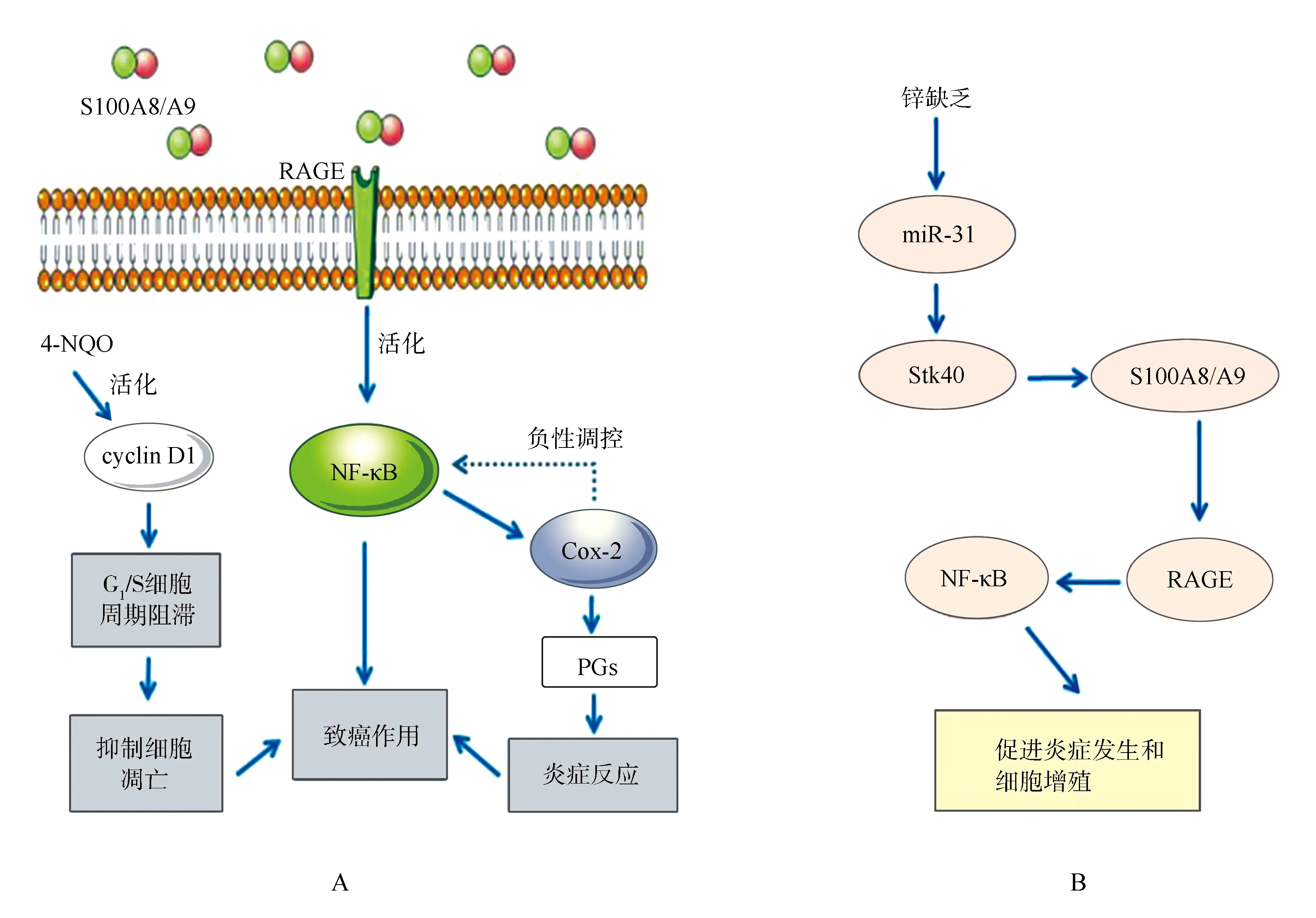
ESCC:食管鳞状细胞癌;RAGE:晚期糖基化终末产物受体;4-NQO:4-硝基喹啉-1-氧化物;cyclin D1:细胞周期蛋白D1;NF-κB:核因子κB;COX-2:环加氧酶2;PGs:前列腺素;miR-31:微RNA-31;Stk40:丝氨酸/苏氨酸激酶40
间变性甲状腺癌(anaplastic thyroid carcinoma,ATC)是TC中最恶性的类型,其特点是早期转移和局部器官浸润[26]。在ATC中,S100A8/A9的表达高于非肿瘤性甲状腺组织和高分化甲状腺肿瘤[27]。S100A8通过与RAGE相互作用激活下游的p38、胞外信号调节激酶1/2(extracellular regulated kinase 1/2,ERK1/2)、c-Jun氨基端激酶(c-Jun N-terminal kinase,JNK)、促分裂原活化的蛋白激酶(mitogen activated protein kinase,MAPK)通路,进而刺激肿瘤细胞生长[27]。通过短发夹技术敲除内源性S100A8,抑制S100A8介导的ATC的体外增殖和体内肿瘤形成,提高动物存活率[27]。S100A8表达的下调能抑制蛋白激酶B的磷酸化,诱导细胞色素C、caspase-3及caspase-9等促凋亡基因表达,进而诱导细胞凋亡[28]。因此,S100A8的靶向性可能有利于ATC患者的预后及治疗[27]。
甲状腺乳头状癌(papillary thyroid carcinoma,PTC)是最常见的TC,预后良好[29]。PTC晚期分泌大量与肿瘤相关的巨噬细胞,这些巨噬细胞是S100A8/A9的主要来源,所以PTC患者的S100A8/A9水平显著升高[6]。氧化应激可导致活性氧类的过度产生或清除不足,这些高活性代谢物通过刺激过氧亚硝酸盐和其他致突变剂导致DNA损伤[6]。S100A8/A9可通过S100A9 C端的HHH结构域与花生四烯酸结合,S100A9在Thr113处的磷酸化可激活还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶产生活性氧类,并激活NF-κB信号通路[1]。有研究表明,PTC患者中的氧化应激指数、血清脂质过氧化物及S100A8/A9水平均呈上升趋势,在甲状腺全切除术后血清S100A8/A9的水平明显下降[6]。提示S100A8/A9可能是这类甲状腺恶性肿瘤的一个有用的生物标志物[6]。另有研究显示,与健康对照组相比,良性甲状腺肿和PTC患者的尿蛋白中S100A8和S100A9蛋白的表达均显著降低[29]。
3 S100A8/A9基因的甲基化与头颈部肿瘤
DNA甲基化、组蛋白修饰等表观遗传改变可在整个细胞生命周期中持续,并遗传给子代细胞[30]。在肿瘤细胞中,常存在正常DNA甲基化模式的异常改变,表现为全基因组广泛低甲基化及抑癌基因CpG岛区域高甲基化[31]。在许多肿瘤中,启动子甲基化导致的基因沉默可能是癌变过程的早期事件,可能较突变和缺失导致的基因结构失活更常见[3]。研究表明,OSCC患者远端正常黏膜组织的平均甲基化水平显著低于OSCC区的甲基化水平,同时显著高于健康供者正常黏膜的甲基化水平[3]。在HNSCC中S100A9表达的下调与增强的甲基化水平呈负相关,而HNSCC中S100A8表达的下调与增强的甲基化可能并不相关[8]。
4 展 望
S100A8/A9在头颈部肿瘤发生发展中发挥了重要作用,有可能成为头颈部肿瘤预防、治疗和预后评定的新指标[1,5-9]。S100A8/A9在头颈部肿瘤发生发展中的作用机制与G2/M期细胞周期阻滞、EGFR异常、RAGE/NF-κB信号通路等密切相关。近年来关于表观遗传调控通过影响S100A8/A9的表达,进而影响头颈部肿瘤发生发展进程的研究逐渐增多[1,30],S100A9在HNSCC中的低表达与增强的DNA甲基化水平呈负相关[8]。随着对S100A8/A9在头颈部肿瘤发生发展中作用机制研究的深入,表观遗传调控在头颈部肿瘤发生发展中的作用也将成为研究的热点。相信随着S100A8/A9的作用及调控机制研究的不断深入,S100A8/A9将会在头颈部肿瘤的预防、诊断及治疗等方面发挥更加重要的作用。
