1例3-甲基戊烯二酸尿症Ⅰ型患儿AUH基因突变检测及分析
2020-12-02王洪芹徐茶宋东坡吕亚囡孙英梅陈艳萍
王洪芹,徐茶,宋东坡,吕亚囡,孙英梅,陈艳萍
1青岛妇女儿童医院,山东青岛266034;2德州市妇幼保健院
3-甲基戊烯二酸(3-MGA)尿症是由于亮氨酸代谢途径异常引起的以尿中3-MGA排出增多为特征的一组遗传代谢病[1]。3-MGA尿症主要包括5种类型[2]:Ⅰ型为AUH基因突变所致3-甲基戊烯二酰辅酶A水解酶(3-MGH)缺乏的常染色体隐性遗传病;Ⅱ型又称Barth综合征,是TAZ基因突变所致线粒体膜蛋白tafazzin功能缺陷的X连锁隐性遗传病;Ⅲ型又称Costeff视神经萎缩综合征,是线粒体膜蛋白OPA3基因突变所致的常染色体隐性遗传病;Ⅳ型为排除其他类型的异常原因的3-MGA尿症[3];Ⅴ型又称扩张型心肌病伴共济失调综合征,为DNAJC19基因突变所致的DNAJC19蛋白改变的常染色体疾病。3-MGA尿症Ⅰ型(MGA1)是一种常染色体隐性遗传病[4],临床罕见,国内尚无相关报道;自1978年以来,只有38例MGA1病例文献报道[1~20]。该病早期可能无症状,因此通过新生儿疾病筛查发现此病尤为重要。2016~2019年,本研究对1例新生儿疾病筛查发现的MGA1患儿及其父母的AUH基因进行检测,应用生物信息学软件预测突变位点致病性和突变可能导致的蛋白质结构及功能的变化。现报告如下。
1 资料与方法
1.1 临床资料 患儿男,3岁,因新生儿疾病筛查串联质谱甲基丙二酰基肉碱(C4DC+C5OH)高于正常参考范围于2016年10月9日来我院就诊。患儿系第2胎,第2产,足月顺产出生,出生体质量为3 600 g,围生期正常。患儿父母健康,否认近亲结婚,无类似疾病家族史,患儿有一同胞姐姐,表型正常。患儿自出生以来,分别于出生时、1个月、3个月、6个月、9个月、1岁、2岁、3岁时进行保健查体,身高、体质量、头围等各项生长发育指标与同龄幼儿无明显差异,无小头畸形,无痉挛性四肢麻痹性精神运动迟缓,无癫痫和肌张力障碍,无语言发育迟缓,无低血糖,无代谢性酸中毒等。实验室检查结果无明显异常。患儿血串联质谱筛查显示,出生1个月、3个月、6个月、9个月、1岁、3岁时C4DC+C5OH分别为1.0、0.8、0.81、1.38、1.74、2.21 μmol/L,明显高于正常参考范围(0.05~0.45 μmol/L)。患儿于1岁时进行尿液有机酸气相色谱-质谱联用分析检测显示,3-羟基异戊(3-HIVA)21.2 mmol/mol肌酐(正常参考范围:0~2.3 mmol/mol肌酐),3-MGA 222.98 mmol/mol肌酐(正常参考范围:0~4.2 mmol/mol肌酐)。其中3-MGA为正常均值的202.71倍(正常均值为1.1 mmol/mol肌酐),初步诊断为3-MGA尿症。
1.2 AUH基因测序及测序验证
1.2.1 高通量测序 征得患儿父母的知情同意,采集患儿及其父母的外周血各2 mL,送杭州甄元医学检验实验室,对致病基因AUH基因的10个外显子进行基因捕获及第二代高通量测序。
1.2.2 Sanger测序验证 用Sanger测序法对患儿及父母AUH基因可疑突变位点进行验证,结果与参考序列比较。
1.3 突变位点生物学信息分析 采用Mutation Taster和PolyPhen-2软件、CRYP-SKIP软件、人类剪接查找服务器(HSF)预测突变位点致病性和突变可能导致的蛋白质结构和功能的变化。Mutation Taster可预测突变位点氨基酸保守性。PolyPhen-2可预测氨基酸替换对人蛋白质结构和功能的影响。CRYP-SKIP隐蔽剪接位点激活-外显子跳跃预测器,可预测因剪接突变导致的隐蔽剪接位点激活(PCR-E)和外显子跳跃(1-PCR-E)的可能性大小,PCR-E的值介于0和1,较高的值表示密码子的隐蔽位点激活,较低的值表示外显子跳跃。HSF可预测隐蔽剪接位点及基因突变对剪接位点的改变[5]。HSF得分范围为0~100,MaxEnt得分范围为-20~20。当突变发生时,若同时满足野生型HSF得分大于65、MaxEnt得分大于3、野生型与突变型之间HSF得分变化率小于-10%、野生型与突变型之间MaxEnt得分变化率小于-30%,则HSF软件预测为该突变破坏了剪接位点。
2 结果
2.1 基因测序及验证结果
2.1.1 高通量测序结果 基因捕获及第二代高通量测序显示患儿AUH基因存在c.894+5G>A杂合突变和c.677G>A杂合突变。c.894+5G>A突变为剪接突变,第894+5号核苷酸由鸟嘌呤变为腺嘌呤,是未报道过的新突变,在ESP6500、千人基因组计划及ExAC数据库中均未检索到。c.677G>A为错义突变,位于第7外显子,第677号核苷酸由鸟嘌呤变为腺嘌呤,在ESP6500、千人基因组计划中也均未检索到,ExAC数据库曾报道过,在dbSNP数据库中检索到其收录ID为rs752044125,为已报道过的突变。
2.1.2 Sanger测序验证结果 Sanger测序验证结果显示患儿母亲携带AUH基因c.894+5G>A剪接突变,患儿父亲携带AUH基因第7外显子c.677G>A错义突变。见图1。患儿的AUH基因c.894+5G>A剪接突变和第7外显子c.677G>A错义突变分别来自于母亲和父亲。
2.2 基因突变致病性预测情况 采用CRYP-SKIP软件和HSF分析c.894+5G>A剪接突变,预测结果为突变使外显子跳跃,其中CRYP-SKIP预测显示,突变后外显子隐蔽剪接位点激活(PCR-E)的可能性为0.08,外显子跳跃(1-PCR-E)的可能性为0.92;经HSF预测软件显示,野生型供体区域的生物保守性HSF得分为89.9,突变后HSF得分为77.73,野生型与突变型之间变异程度为-13.54%,5′剪接位点野生型MaxEnt得分为9.65,野生型与突变型之间变化率为-37.41%;由以上数据可知,此突变一定程度上破坏了相邻第8外显子的供体区域,影响剪接。采用Mutation Taster、PolyPhen-2软件对c.677G>A错义突变进行生物信息学分析,被替代的碱基为高度保守序列(图2),此突变可能致病;PolyPhen-2软件对c.677G>A错义突变预测得分为1,得分越接近1,突变越可能导致蛋白功能改变而致病。
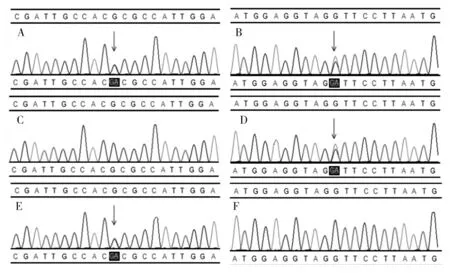
注:A为患儿c.677G>A突变;B为患儿c.894+5G>A突变;C为患儿母亲未测出c.677G>A突变;D为患儿母亲c.894+5G>A突变;E为患儿父亲c.677G>A突变;F为患儿父亲未检测到c.894+5G>A突变。

图2 Mutation Taster软件分析c.677G>A突变位点保守性结果
3 讨论
3-MGA尿症是一组以尿中3-MGA排出增多为特征的遗传代谢病。正常人尿液中3-MGA含量小于20 mmol/mol肌酐,而3-MGA尿症患者尿中3-MGA含量可间歇性超过1 000 mmol/mol肌酐[3,6]。本例患儿属于MGA1,尿中3-MGA明显增高,为正常均值的202.71倍。研究显示,3-MGH在小鼠肾脏、骨骼肌、心脏和大脑中高度表达,定位于线粒体内[7]。3-MGH具有RNA结合功能和烯酰辅酶A水合酶活性两种功能。甲基戊二酰辅酶A水合酶催化水合作用,将3-甲基戊烯二酰辅酶A催化形成3-羟基-3甲基戊二酸,这是亮氨酸降解途径中的关键步骤。3-MGA尿症主要是由于3-MGH缺乏,使3-甲基戊烯二酰辅酶A不能水解为3-羟基-3甲基戊二酸,导致前体代谢产物3-MGA和3-HIVA排出增多[8]。通常,MGA1患者尿中3-MGA水平高于其他几种类型,此外,MGA1患者尿中3-HIVA排泄增加,可以此作为区别3-MGH缺乏症和其他类型3-MGA尿症的一个重要参数[9]。MGA1患者尿液中3-MGA顺式和反式两种异构体的比例为2∶1,脑脊液中只有顺式异构体,而在其他类型3-MGA尿症中,尿液顺式和反式3-MGA比例约为1∶1[10]。亮氨酸负荷试验可以鉴别由AUH基因突变导致的MGA1[11]。
3-MGH由AUH基因编码,该基因定位于9号染色体9q22.31,包含10个外显子,编码339个氨基酸[12]。目前,文献中已有11种不同的突变被报道,其中最常见的突变类型是错义和无义突变[1]。经预测,这其中许多突变将导致蛋白质功能的完全丧失[13]。本例患儿未表现出3-MGA尿症相关症状,为出生后进行新生儿疾病筛查串联质谱检测C4DC+C5OH高于正常参考范围所发现,随后进行患儿尿液分析,符合3-MGA尿症的诊断,基因检测显示存在AUH基因c.894+5G>A突变和第7外显子c.677G>A复合杂合突变。经Sanger测序验证,c.894+5G>A剪接突变来自母亲,c.677G>A错义突变来自父亲。通过CRYP-SKIP软件和HSF对c.894+5G>突变预测,发现在外显子-内含子结合点(5′剪接位点)发生突变,此突变一定程度上破坏了相邻第8外显子的供体区域,导致外显子跳跃,造成剪切位点改变,影响正常RNA在该位点的剪接,推测可能导致编码蛋白的结构改变从而影响蛋白功能。通过Mutation Taster、PolyPhen-2软件对c.677G>A错义突变进行预测,发现被替代的碱基为高度保守序列,c.677G>A突变导致第226位精氨酸被组氨酸替代,蛋白质发生p.R226H改变,可能导致蛋白质结构和功能改变,从而可能致病。然而,上述2种基因新突变的致病性仅通过软件预测得出,尚需体外蛋白功能表达研究进行证实。
MGA1的发病起始年龄可从婴儿期到成人[4]。该型临床表现差异较大,可从无症状到广泛的轻度异质性神经功能障碍、语言发育迟缓、四肢瘫痪、肌张力障碍、舞蹈动作、严重脑病、精神运动迟缓、婴儿期或儿童期基底神经节受累和成人脑MRI示脑白质脑病变、共济失调和痉挛状态[1]。2008年Leipnitz等[14]研究发现,3-MGA的毒性影响仅作用于大鼠的大脑皮层,3-HIVA可聚集在患者的脑脊液中,具有神经毒性。2010年Wortmann等[15]曾报道过2例儿童MAG1患者MRI显示有基底神经节病变和脑萎缩,而另外2例患者脑部MRI显示正常。MGA1是一种典型的有机酸尿症,目前没有针对该病的特异性治疗方法。由于MGA1患者在进行高亮氨酸饮食或高蛋白质饮食后尿液中3-MGA含量会明显增多,因此,目前比较常用的治疗方法为限制亮氨酸或蛋白质饮食[16,17]。本例患儿自出生以来,常规儿童保健查体与同龄儿童相比未见明显异常,建议进一步行脑MRI检查,排除脑白质病变。由于该病发病起始年龄不定且临床表现差异大,建议患儿定期检查,长期随访。
综上所述,本例3-MGA尿症患儿通过高通量测序及Sanger验证,检测到患儿AUH基因c.894+5G>A和c.677G>A复合杂合突变。经软件预测分析,此2种突变很有可能致病,为该患儿的遗传学病因。其中,c.894+5G>A剪接突变是未报道过的新突变,c.677G>A错义突变为已报道过的突变。本例c.894+5G>A剪接突变和c.677G>A错义突变的检出丰富了AUH基因谱,为3-MGA尿症的诊断和产前咨询提供了依据。
