Pt/WC催化剂甲醇脱氢反应机理的研究
2020-11-30刘婷王巍施静敏
刘婷 王巍 施静敏


摘 要: 采用密度泛函理论(DFT)和周期平板模型,研究了甲醇在PtML/WC(0001)催化剂表面上的脱氢反应机理。通过对甲醇分解过程中反应物、中间体及产物的结构优化和对可能基元反应的过渡态搜索,得到了各吸附物种在最稳定吸附构型下的吸附能和各基元反应的活化能垒。通过对三条可能反应路径的比较,即甲醇分子中O—H键断裂生成甲氧基(CH3O*)和氢(H*)、C—H键断裂生成羟甲基(CH2OH*)和氢(H*)、C—O键断裂生成甲基(CH3*)和羟基(OH*),发现O—H键的断裂所需活化能最低。因此,在PtML/WC(0001)表面上,甲醇分子中的 O—H键断裂生成CH3O*为整个反应的速控步骤。
关 键 词:密度泛函理论;Pt/WC催化剂;甲醇脱氢;反应机理
中图分类号:O641;O647 文献标识码: A 文章编号: 1671-0460(2020)09-1941-04
Abstract: The density functional theory (DFT) and periodic slab models were used to investigate the reaction mechanism of methanol dehydrogenation on the PtML/WC(0001) catalyst surface. The geometrical structures of reactants, intermediates and products involved in the methanol decomposition were optimized and the transition states of the possible elementary reactions were searched. As a result, the adsorption energies of possible adsorbed species with the most stable configurations and the activation energy barriers of the possible elementary reactions were obtained. Three possible reaction pathways, including the O—H bond breaking in methanol to form methoxy(CH3O*) and hydrogen(H*), the C—H bond breaking to form hydroxymethyl(CH2OH*) and hydrogen(H*), and the C—O bond breaking to form methyl(CH3*) and hydroxyl(OH*), were compared. It was found that the O–H bond breaking pathway had the lowest energy barrier. Thus, the O—H bond breaking in the methanol molecule to CH3O* is the rate-limiting step in the process of methanol dehydrogenation.
Key words: Density functional theory ; Pt/WC catalyst ; Methanol dehydrogenation ; Reaction mechanism
伴隨我国环境污染日益严重,清洁能源的发展受到极大关注。铂(Pt)系贵金属催化剂是电解析氢催化活性较高的催化剂,但由于价格昂贵等因素制约了其商业化应用。自发现碳化钨(WC)具有类Pt催化活性后,廉价的过渡金属碳化物的催化性能受到关注[1]。作为电催化剂,虽然WC具有优良的抗CO中毒性能及电化学稳定性,但其电催化活性却不 高[2]。近来,碳化钨负载微量铂(Pt/WC)的复合型催化材料凭借其良好电催化活性引起了实验工作者的极大兴趣[3-5]。密度泛函理论(DFT)是催化剂表面上研究催化反应性质的必要手段,单一过渡金属和贵金属表面上的催化反应机理已有大量研究,不同金属表面上,反应机理及速控步骤均有所不同。因此,本文采用DFT方法在Pt原子单层(ML)和WC(0001)催化剂表面上对甲醇的脱氢反应历程进行了较详尽的讨论,为Pt/WC复合催化剂催化甲醇分解反应的研究提供理论依据。
1 计算模型与方法
1.1 计算模型
WC具有六方紧密堆积结构,属(P-6m2)对称点群。WC (0001)表面具有与Pt、Pd等相似的催化活性,因此成为实验和理论重点研究的表面[6-7]。 本文对甲醇脱氢反应历程的模拟基于此表面进行,如图1所示。选取有周期性边界条件的PtML/WC (0001)-2×2五层平板模型进行甲醇脱氢反应计算。相邻两平板间的真空层厚度为1 nm,以确保分子间相互作用力足够小。PtML/WC(0001)复合表面的吸附位包括: top、桥位(bridge)、fcc和hcp穴位(如图1所示)。
1.2 计算方法
全部工作采用Materials Studio软件包中基于密度泛函理论[8-9]的CASTEP[10]模块完成。交换相关势采用广义梯度积分(GGA)和Perdew-Wang-91 泛函相结合的方法[11],离子实与价电子之间的相互作用采用超软赝势(ultrasoftpsedupotential) [12],平面波基组的动能截断值为300 eV,表面布里渊区域的K格点为4×4×1。用上述方法计算得到WC的晶胞结构参数为 a=0.293 4,c=0.286 2 nm,与实验值 (a= 0.290 6,c=0.283 7 nm)[13]相吻合,说明本文采用的计算方法是可靠的。过渡态计算使用完全线性同步和二次同步变换(complete LST/QST)方法[14]。
甲醇脱氢反应中涉及的反应物及中间产物的吸附能()定义为:
2 结果与分析
2.1 各吸附物种的结构和能量
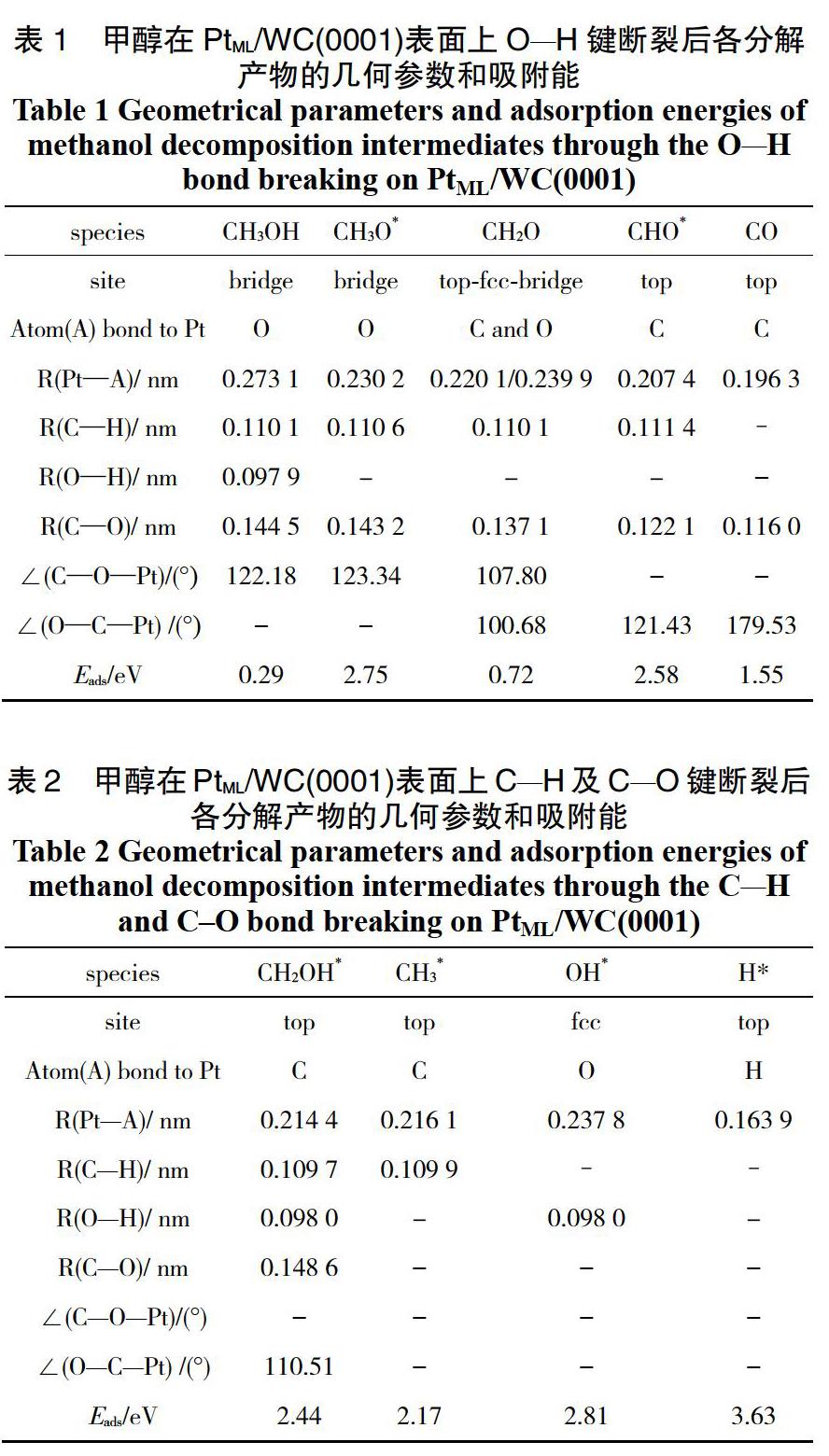
吸附物种在表面上的平衡几何构型对于表面催化反应的研究至关重要。通过对甲醇及其脱氢反应过程中可能涉及的中间产物进行结构优化,得到了各吸附物种在PtML/WC(0001)表面最稳定的吸附构型和相应的吸附能,结果列于表1和表2。
2.1.1 甲醇(CH3OH)
CH3OH分子在PtML/WC(0001)复合表面上最稳定的吸附构型是通过O原子吸附于桥位,如图2(a)所示。CH3OH 的O原子与表面Pt原子间距为 0.273 1 nm, 考虑到O和Pt原子的范德华半径分别为0.152 nm和0.175 nm[15],所以Pt—O间距小于O和Pt原子的范德华半径之和(0.327 nm),说明CH3OH分子与表面发生相互作用。经计算得到自由CH3OH分子的C—O键长为0.143 7 nm,O—H键长为 0.096 8 nm,这与实验值[16] (C—O键长为0.142 5 nm,O—H键长为0.094 5 nm)吻合较好。CH3OH分子的吸附能仅为0.29 eV,进一步说明CH3OH与该表面存在较弱相互作用。
2.1.2 甲氧基(CH3O*)
此中间体在复合表面上最稳定吸附构型是通过O原子吸附于Pt—Pt桥位,并且CH3O*中C—O键轴与表面成46.10°倾斜角,如图2(b)。CH3O*的O原子与表面Pt原子形成的两个Pt—O键长为 0.230 2 nm,与吸附CH3OH分子中Pt—O键长 (0.273 1 nm)相比缩短了0.042 9 nm。相应的CH3O*吸附能比CH3OH增加了2.46 eV,这表明CH3O*在此表面上是很强的化学吸附。
2.1.3 甲醛(CH2O)
CH2O在表面上通过C和O原子吸附于top-fcc-bridge位,且CH2O中C—O键轴与表面几乎平行, 如图2(c)。吸附后CH2O分子中C—O键长(0.137 1 nm)与计算得到的自由CH2O分子C—O键长(0.122 3 nm)相比,有明显拉长。说明吸附后CH2O分子中C=O双键变弱,趋向于单键。CH2O分子在表面的吸附能与CH3OH分子相比,明显增加,分子与表面相互作用增强。
2.1.4 甲酸基(CHO*)
CHO*通过C原子吸附于表面顶位(图2(d)),O—C—Pt键角为121.43°。在此最稳定构型中,C—O键长为0.122 1 nm,与吸附后CH2O分子中C—O键长相比明显缩短,说明CHO*中C=O键为双键。CHO*在表面上吸附能为2.58 eV,说明该中间体与金属表面具有很强相互作用。
2.1.5 一氧化碳(CO)
CO分子在表面上最稳定构型是通过C原子吸附于顶位,如图2(e)。 O—C—Pt键角为179.53°,表明CO中C—O键轴与表面垂直。CO中C原子与表面Pt原子间距0.196 3 nm,吸附能1.55 eV。 MILLINGER[17]等通过TPD实验研究了Pt/WC多晶复合薄膜上饱和CO覆盖度下CO与表面的结合能。结果表明,在0.8 和2 ML Pt修饰WC表面上,CO结合能分别为94.5和102.1 kJ?mol-1 (即0.98和 1.05 eV)。
由于CO的吸附能随覆盖度增大而减小,因此Millinger等在饱和CO覆盖度下得到的实验值会略小于本文在0.25 ML CO覆盖度下的计算结果,这表明本文计算结果非常可靠。另外,在相同条件下计算了Pt(111)表面上CO吸附能。结果发现,CO在Pt(111)表面吸附能为1.74 eV。这意味着Pt/WC复合催化剂与传统Pt催化剂相比,其对甲醇氧化过程中产生的中间产物CO吸附能减小,抗CO中毒能力提高。
2.1.6 羟甲基(CH2OH*)、甲基(CH3*)、羟基(OH*)和氢原子(H*)
CH2OH*在复合表面上最稳定吸附构型是通过C原子吸附于顶位(图2(f)),其吸附能为2.44 eV。與CH3OH中C—O键长相比,CH2OH*的C—O键拉长了0.004 1 nm,这表明通过O原子与表面吸附的能力(如CH3OH)弱于通过C原子与表面吸附(如CH2OH*)。
CH3*在PtML/WC(0001)表面上最稳定的吸附构型是通过C原子吸附于顶位(图2(g))。此构型中,CH3*保持C3ν对称性,CH3*中3个H原子所在平面与表面平行。其C原子与表面Pt原子的距离为0.216 1 nm, 吸附能为2.17 eV。OH*在复合表面上通过O原子吸附于三重fcc穴位,且O—H键轴垂直于表面,如图2(h)所示。
H*在复合表面上最稳定吸附位是顶位(图2(i)),吸附能高达3.63 eV。Pt—H键长为0.163 9 nm,远小于H和Pt原子的范德华半径之和(0.295 nm)[15],说明H*在金属表面上发生很强化学吸附。
由上看出,自由基物种吸附能力始终强于分子物种吸附能力。这是由于自由基中间体存在未成对电子,使自由基活性增加,在与金属表面发生吸附的过程中有更强接受电子的能力。并且在不受空间位阻因素的影响下,各物种倾向于通过C原子而不是O原子与金属表面发生相互作用。
2.2 反应机理
甲醇脱氢分解反应是一个涉及多个中间体的复杂反应。甲醇分解开始于自由甲醇分子在PtML/WC(0001)表面发生吸附。吸附后的甲醇分子通过一系列连续脱氢步骤生成反应中间体。甲醇分子中包括三种不同成键, 即C–H、C–O和O–H键。因此,甲醇分解的第一步即为甲醇分子的C–H、C–O或者O–H键活化解离,以引发催化分解反应进行。上述步骤的反应方程式如下:
CH3OH分子中C—H、C—O和O—H鍵的活化解离过程及相应过渡态和产物的构型示于图3。CH3OH分子中O—H键的断裂将产生中间体产物CH3O*和H*。在O—H键的活化解离反应中,将CH3O*和H*置于相邻的最稳定吸附位构成的共吸附体系进行构型优化,优化后共吸附体系与其他吸附体系相比稳定。因此,将其作为反应终态,进行过渡态搜索,过渡态结构示于图3(TS1)。与初始态吸附的CH3OH分子相比, TS1中O—H键拉长了0.038 9 nm,O原子与表面Pt原子间距缩短了0.029 7 nm,O—H键中H原子与表面Pt原子间距由0.322 3 nm 缩短到0.188 9 nm。这说明CH3OH中O–H键被活化,O—H键成键作用减弱, Pt与H间成键作用增强。此过程活化能垒是1.08 eV,为吸热反应,所需热量0.19 eV。
CH3OH分子中C—H键的断裂产生中间体CH2OH*和H*。与O—H键的解离反应类似,终态为CH2OH*和H*置于相邻最稳定吸附位。过渡态结构示于图3(TS2)。与初态相比,TS2中C和H原子间距由0.110 1 nm 拉长到0.192 4 nm,H和Pt原子间距由0.262 4 nm 缩短到0.171 2 nm。说明H原子在表面与Pt原子趋于成键,促使C—H键断裂。此反应的活化能为1.47 eV,反应放热0.04 eV。
CH3OH分子中C—O键的断裂产生中间体CH3*和OH*,过渡态结构示于图3(TS3)。在TS3构型中,C—O键由0.144 5 nm拉长为0.227 9 nm,O原子与表面Pt原子间距由0.273 1 nm缩短到0.242 6 nm,表明C—O键发生解离。此过程的活化能垒高达 1.87 eV,反应放热0.19 eV。
由上可知,尽管CH3OH分子中O—H键的解离为吸热反应,但其具有最低活化能垒,因而使O—H键最容易发生解离。由此可以判定CH3OH分子在PtML/WC(0001)表面上的分解反应是从O—H键解离引发的。
3 结 论
采用密度泛函理论,对PtML/WC(0001)复合表面上甲醇脱氢的反应机理进行了研究。通过对甲醇脱氢过程中所涉及反应物、中间体和产物进行几何构型优化,并对各基元反应进行过渡态搜索,得到了各物种在复合表面上最稳定的吸附构型和吸附能以及各反应的活化能垒。
结果表明,CH3OH、CH3O*、CH2O、CHO*、CO、CH2OH*、CH3*、OH*和H*在复合表面的吸附能分别是0.29、2.75、0.72、2.58、1.55、2.44、2.17、2.81、3.63 eV,分子物种的吸附弱于自由基物种。在不受空间位阻因素的影响下,各物种倾向于通过C原子而不是O原子与金属表面发生相互作用。甲醇在PtML/WC(0001)表面上的分解主要经历O—H键断裂生成甲氧基CH3O*中间体,所需活化能为1.08 eV,为整个反应的速控步骤。
参考文献:
[1]LEVY R B, BOUDART M. Platinum-like behavior of tungsten carbide in surface catalysis [J]. Science, 1973, 181(4099): 547-549.
[2]ZELLNER M B, CHEN J G. Surface science and electrochemical studies of WC and W2CPVD films as potential electrocatalysts [J]. Catal. Today, 2005, 99(3-4):299-307.
[3]ZHONG S W, HU X C, YU Y, et al. Core- shell hierarchical tungsten carbide composite microspheres towards methanol electrooxidation [J]. J. Fuel Chem. Tech.,2018, 46(5):585-590.
[4]LIU Y, YOU H, KIMMEL Y C, et al. Self-terminating electrodeposition of Pt on WC electrocatalysts[J]. Appl. Surf. Sci.,2020, 504: 144472-144477.
[5]杨翩翩,黄丽珍,李影影,等. 掺氮碳化钨的制备及其电催化性能的研究[J].电化学,2018,24(1):63-71.
[6]HOUSTON J E, LARAMORE G E, PARK R L. Surface electronic properties of tungsten, tungsten carbide, and platinum [J]. Science, 1974, 185(4147):258-260.
[7]赵学华,王俊文,王晓斌,等. 纳米WC 粉体的制备及其催化应用 [J].粉末冶金技术,2012,30(4):307-312.
[8]刘存海,孙江,柳叶. 路易氏气的密度泛函理论研究[J].当代化工,2018,47(10):2082-2084.
[9]HOHENBERG P, KOHN W. Inhomogeneous electron gas [J]. Phys. Rev. B, 1964, 136:864-871.
[10]SEGALL M D,LNDAN P J D, PROBERT M J, et al. First principles simulation: ideas, illustrations and the CASTEP code [J]. J. Phys. Condens. Matter., 2002, 14:2717-2744.
[11]PERDEW J P, CHEVARY J A, VOSKO S H, et al. Atoms, molecules, solids, and surfaces:applications of the generalized gradient approximation for exchange and correlation [J]. Phys. Rev. B, 1992, 46:6671-6687.
[12]VANDERBILT D. Soft Self-consistent pseudopotentials in a generalized eigenvalue formalism [J]. Phys. Rev. B, 1990, 41(11):7892-7895.
[13]SUETIN D V, SHEIN I R,IVANOVSKII A L. Structural, electronic properties and stability of tungsten mono- and semi-carbides :a first principles investigation [J]. J. Phys. Chem. Solids, 2009, 70: 64-71.
[14]PANG X, YANG J, PANG M, et al. Adsorption and migration behavior of molybdenum atom on graphite (0001) surface [J]. Appl. Surf. Sci., 2019, 470:1064-1070.
[15] BONDI A. Van der Waals' volumes and radii [J]. J.Phys.Chem., 1964, 68:441-451.
[16]LIDE D R. CRC handbook of chemistry and physics 81th Ed[M].. BocaRaton:CRC Press, 2000.
[17]MELLINGER Z J,WEIGERT E C,STOTTLEMYER A L, et al. Enhancing CO tolerance of electrocatalysts:electro-oxidation of CO on WC and Pt-modified WC [J]. Electrochem. Solid-State Lett., 2008, 11(5): B63-B67.
