Candidate genes associated with cold-coagulation or heataccumulation blood stasis syndrome in hypertension
2020-11-27RuiXueChenMeiXiaFangQiuErLiangJingZhiZhangDepartmentofTraditionalChineseMedicineInstituteofIntegrationofTraditionalandWesternMedicineStateKeyLaboratoryofRespiratoryDiseasetheSecondAffiliatedHospitalGuangzhouMedicalUnive
Rui-Xue Chen,Mei-Xia Fang,Qiu-Er Liang,Jing-Zhi ZhangDepartment of Traditional Chinese Medicine,Institute of Integration of Traditional and Western Medicine,State Key Laboratory of Respiratory Disease,the Second Affiliated Hospital,Guangzhou Medical University,Guangzhou 5060,China;Guangzhou Institute of Cardiovascular Disease,Guangdong Key Laboratory of Vascular Diseases,the Second Affiliated Hospital,Guangzhou Medical University,Guangzhou 5060,China;Institute of Laboratory Animals,Jinan University,Guangzhou 506,China;School of Chinese Medicine,Jinan University,Guangzhou 506,China.
Abstract Background:The goal of this study was to predict candidate genes by analyzing the differentially expressed genes of cold-coagulation or heat-accumulation blood stasis syndrome in hypertension by transcriptomes sequencing in human vascular endothelial cells models.Methods:Serum of patients with hypertension were collected to incubate with normal human umbilical vein endothelial cells to establish injured endothelial cell models of cold-coagulation blood stasis syndrome,heat-accumulation blood stasis syndrome and non-blood stasis syndrome.The differentially expressed genes of cold-coagulation blood stasis syndrome or heat-accumulation blood stasis syndrome were screened compared with non-blood stasis syndrome.Gene Ontology,pathway enrichment analyses and PPI network analyses were conducted to get the key genes of cold-coagulation blood stasis syndrome or heataccumulation blood stasis syndrome.Results: The results showed that compared with non-blood stasis syndrome,there were 368 differentially expressed genes in cold-coagulation blood stasis syndrome (275 up-regulated,93 down-regulated),and 271 differentially expressed genes in heat-accumulation blood stasis syndrome (202 upregulated,69 down-regulated).According to the bioinformatics analyses,5 differentially expressed genes were selected as the candidate genes for cold-coagulation blood stasis syndrome (TRIB3,HERPUD1,ERN1,PMAIP1 and XBP1) and 10 differentially expressed genes were selected as the candidate genes for heat-accumulation blood stasis syndrome (PTGS2,SLC3A2,VEGFA,SLC7A5,SLC1A4,SLC7A1,SLC38A1,SLC43A2,HMOX1 and ICAM1).Conclusion: In this study,we predicted the potential key genes associated with cold-coagulation blood stasis syndrome or heat-accumulation blood stasis syndrome in hypertension by transcriptomes sequencing and bioinformatics analyses.It provide an informative basis for studying the role of genes in blood stasis syndrome,and we hope it will be valuable to study blood stasis syndrome in future studies.
Keywords:RNA sequencing,Cold-coagulation blood stasis,Heat-accumulation blood stasis,Hypertension
Introduction
In traditional Chinese medicine,blood stasis syndrome(BSS) refers to blood stagnation or coagulation in blood vessels,of which the clinical symptoms are manifested in multiple systems and tissues.BSS has been proved to be a basic pathogenic factor in many diseases,such as hypertension,coronary heart disease,diabetes mellitus,etc [1-3].It was mainly caused by the increase of blood viscosity,platelet adhesion rate and aggregation,or the change of microcirculation,which can accelerate the course of disease [4,5].Therefore,early diagnosis and treatment of BSS may be useful to alleviate the occurrence and development of diseases.
Studies on the mechanism of BSS are complicated.The occurrence of BSS has been closely associated with endothelial dysfunction in many diseases [6,7].Modern pathological studies have proven that vascular endothelial dysfunction in coronary and hypertension was an early marker of the development of atherosclerotic changes and can contribute to cardiovascular events [8].Genetic materials could regulate and change the vascular endothelial functions which may participate in the development of hypertension [9-11].According to clinical features,BSS was often divided into different types,including Qi-stagnation BBS,Qi-deficiency BBS,coldcoagulation BBS (CCBS),heat-accumulation BBS(HABS),etc.It was known that hypertension and BSS were closely associated with vascular endothelial dysfunction,so it was a good research strategy to combine them.
In the previous studies,we analyzed miRNAs levels in endothelial cells exposed to hypertensive patients’serum and found some candidate miRNAs [12].In this study,we collected patients diagnosed as hypertension with CCBS,HABS or non-BBS (NBS).Then,vascular endothelial cells were incubated with patients’ serum to establish the injured endothelial cell models of CCBS,HABS and NBS.RNA sequencing were conducted and the differentially expressed genes(DEGs) were screened.Finally,the candidate genes were chosen by bioinformatics analyses,which might be used as biomarkers or clinical diagnostic targets for CCBS or HABS in hypertension.
Methods
Patients’serum
Patients with hypertension were diagnosed according to the “1999 World Health Organization-International Society of Hypertension Guidelines for the management of hypertension” [13].Systolic blood pressure (SBP) ≥ 140 mmHg or diastolic blood pressure (DBP) ≥90 mmHg could be diagnosed as hypertension.
CCBS or HABS were diagnosed according to the“Chinese Association of the Integration of Traditional and Western Medicine Consensus of Integrative Medicine on BSS Diagnosis and Treatment in 2011”[14].The typical symptoms of CCBS and HABS were as follows.
CCBS:intolerance to cold and cold limbs,peripheral coldness,exacerbated by exposure to cold,pale face,pale tongue with white fur,deep pulse,tight pulse,slow pulse,or taut pulse.
HABS:fever,ozostomia,bitter taste,xerostomia,astriction or yellow urine,dark red tongue with yellow thick fur,rapid pulse,or slippery pulse.
Fasting venous blood was collected,placed at room temperature for 30 minutes and centrifuged at 4 °C,2,000 revolutions per minute for 15 minutes.The serums were taken into sterile EP tubes,inactivated at 56 °C for 30 minutes and stored at -20 °C for reserve.Samples in each group were mixed in equal volume before use.
Establishing Cell Models
HUVECs (ATCC,USA)were cultured in culture flasks(1 × 105* mL-1,25 mL) in Dulbecco’s modified Eagle’s medium (Lot.8119052,Gibco,USA)containing 10% fetal bovine serum (Lot.1989478,Gibco,USA) for 24 hours (37 °C,5% CO2).Then washed the cells,and added a 10% serum of patients with CCBS,HABS or NBS to culture flasks for 24 hours.Then,the injured endothelial cell models of CCBS,HABS and NBS were established.Then cells were digested with TRIzol (Lot.103103,Gibco,USA)and collected into cryopreservation tubes and stored at-80°C.
mRNA library construction and sequencing
The mRNA library was constructed by Illumina Truseq RNA (300 bp).Hiseq2000 platform was used for 2*100 bp sequencing to complete human mRNA sequencing.Quality control of the raw reads was conducted to get high quality and clean reads.The TopHat software (http://tophat.cbcb.umd.edu/) was used to map the high quality and clean reads to the human reference genome.Then the Cuffdiff software(http://cufflinks.cbcb.umd.edu/) was used to calculate the fragments per kilobase of exon model per million mapped reads values of genes in the samples.Finally,the DEGs between samples were selected (FC >1,Pvalue <0.05).
DEGs screening
The DEGs of CCBS were screened compared with NBS.And the DEGs of HABS were screened compared with NBS.Then the DEGs associated with CCBS or HABS were selected for the following bioinformatics analyses.
Gene Ontology(GO)and pathway enrichment analyses
The DEGs were analyzed by DAVID(https://david.ncifcrf.gov/) for GO and Kyoto Encyclopedia of Genes and Genomes (KEGG)pathway significance enrichment analyses (Pvalue <0.05).GO analysis could classify genes by biological process (BP),molecular function and cellular component.
GO enrichment analyses explained the functional enrichment of genes.Pathway enrichment analyses annotated the function of a set of genes instead of single gene.Enrichment analyses could improve the reliability of research.
Protein-protein interactions (PPI) network analyses
The DEGs were mapped to STRING 11.0(https://string-db.org) and imported into the Cytoscape software to obtain the PPI network.The most significant module in the PPI networks was identified by molecular complex detection,which is for clustering a network based on topology to find densely connected regions.Then the genes in this module were selected as the hub genes for the following analyses.
Candidate genes
The candidate genes of CCBS or HABS were chosen from the hub genes of PPI network modules,and the significant enrichment of GO annotations of CCBS or HABS should be satisfied meanwhile (-Log10(Pvalue >5)).
Results
Patients’characteristics
32 cases of hypertension were enrolled from the Second affiliated hospital of Guangzhou Medical University (Guangzhou,China),including 10 cases of CCBS,10 cases of HABS and 12 cases of NBS.Patients with interventional therapy or surgical operation should be excluded.The participants in this study received a complete explanation of the protocol and signed the consent form.The ethical approvals for the study were permitted by the Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University (Approval No.2013024).The characteristics of the patients with BSS compared with NBS were as follows.There was no significant difference between the two groups (Pvalue >0.05)(Table 1).
MRNA sequencing
Clean reads of genes were obtained after quality control and mapped to the human reference genome by Bowtie software(Table 2).
DEGs related to CCBS or HABS
The number of DEGs between groups were analyzed.Compared with NBS,the number of DEGs in CCBS was 368 (275 up-regulated,93 down-regulated).Compared with NBS,the number of DEGs in HABS was 271(202 up-regulated,69 down-regulated).
GO and pathway enrichment analyses
GO enrichment analyses showed that the DEGs of CCBS were mainly enriched in BP,such as intrinsic apoptotic signaling pathway in response to endoplasmic reticulum stress (n=10),cellular response to glucose starvation (n=9),response to endoplasmic reticulum stress (n=10),amino acid transport(n=7),cellular response to hypoxia(n=10),etc (Figure 1A).KEGG pathway enrichment analyses showed that the DEGs of CCBS primarily active in HIF-1 signaling pathway (n=9),p53 signaling pathway (n=7),TNF signaling pathway (n=8),etc(Figure 1B).
GO enrichment analyses showed that the DEGs of HABS were mainly enriched in BP,such as cellular response to hypoxia (n=12),intrinsic apoptotic signaling pathway in response to endoplasmic reticulum stress (n=7),amino acid transport (n=7),positive regulation of apoptotic process (n=15),etc.And in molecular function,genes were mainly enriched in amino acid transmembrane transporter activity (n=6),etc.In cellular component,genes were mainly enriched in cytoplasm(n=91),cytosol(n=64),etc (Figure 1C).KEGG pathway enrichment analyses showed that the DEGs of HABS primarily active in TNF signaling pathway (n=7),p53 signaling pathway(n=5),MicroRNAs in cancer(n=10),etc(Figure 1D).
PPI network analyses
PPI network analyses of the DEGs were conducted by STRING and Cytoscape,and the most significant module in the PPI networks were selected by molecular complex detection.PPI network analyses showed that a total of 23 genes were identified as hub genes in the module in CCBS (Figure 2A).And a total of 15 genes were identified as hub genes in the module in HABS(Figure 2B).
Candidate genes
There were 18 DEGs of CCBS in the significantly enriched GO annotations (-Log10(Pvalue >5)).Then 5 hub genes were chosen as the candidate genes from the 18 DEGs as the potential key genes of CCBS,including TRIB3,HERPUD1,ERN1,PMAIP1 and XBP1(Table 3).
There were 24 DEGs of HABS in the significantly enriched GO annotations (-Log10(Pvalue >5)).And 10 hub genes were chosen as the candidate genes from the 24 DEGs as the potential key genes of HABS,including PTGS2,SLC3A2,VEGFA,SLC7A5,SLC1A4,SLC7A1,SLC38A1,SLC43A2,HMOX1 and ICAM1(Table 4).
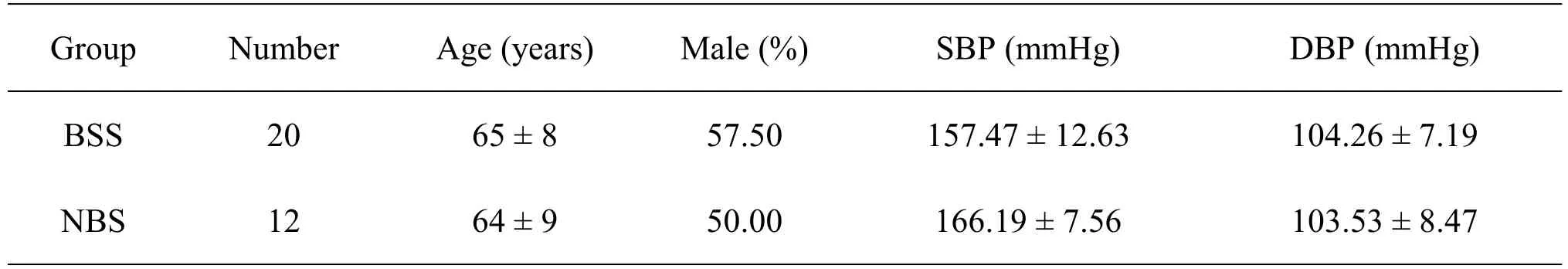
Table 1 The characteristics of the patients
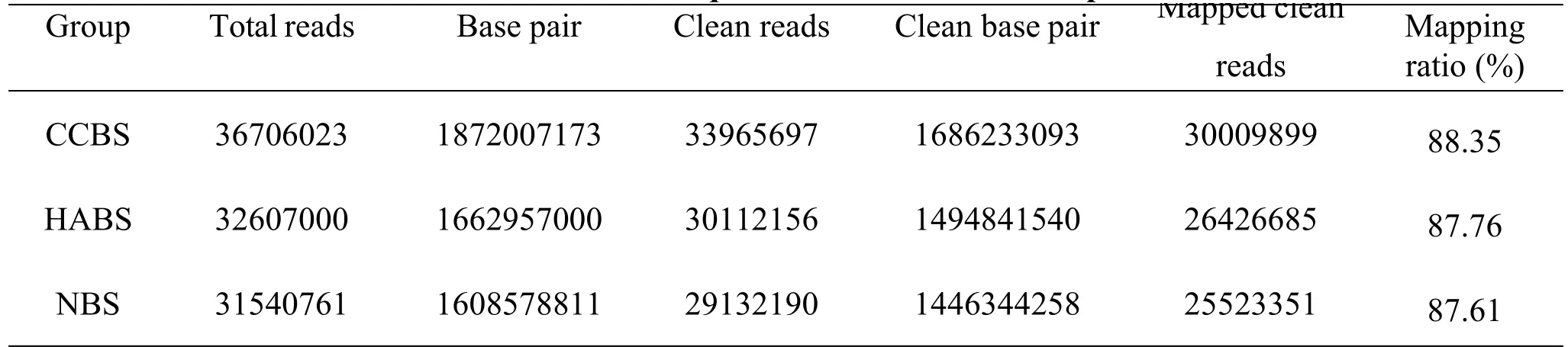
Table 2 mRNAs sequence statistics of the samples
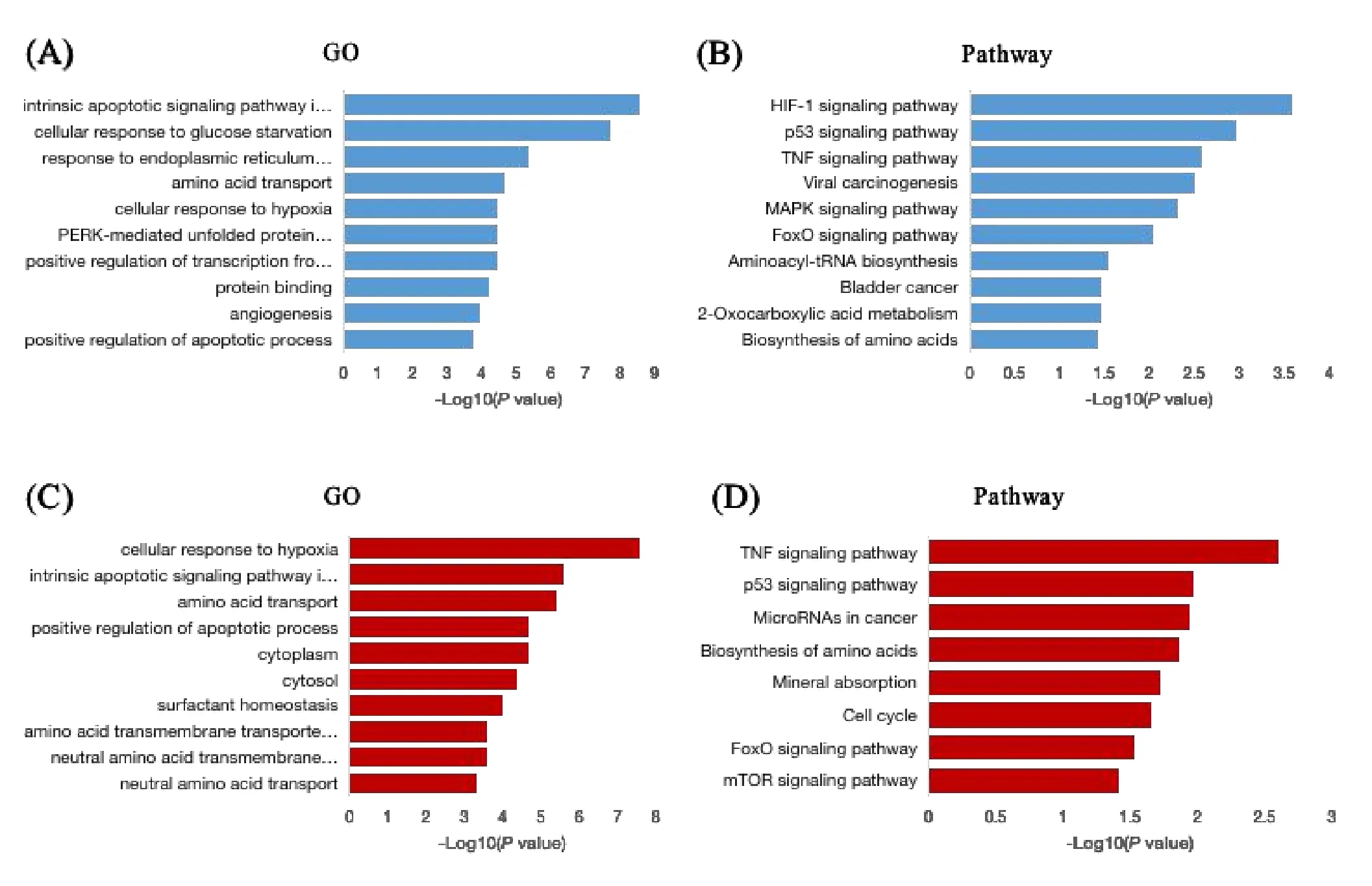
Figure 1 The DEGs in CCBS or HABS compared with NBS analyzed by DAVID software.(A)GO enrichment analysis of the DEGs in CCBS;(B) KEGG Pathway enrichment analysis of the DEGs in CCBS;(C) GO enrichment analysis of the DEGs in HABS;(D) KEGG pathway enrichment analysis of the DEGs in HABS.BSS,blood stasis syndrome;CCBS,cold-coagulation BBS;DEGs,differentially expressed genes;KEGG,Kyoto Encyclopedia of Genes and Genomes;HABS,heat-accumulation BBS;GO,Gene Ontology.
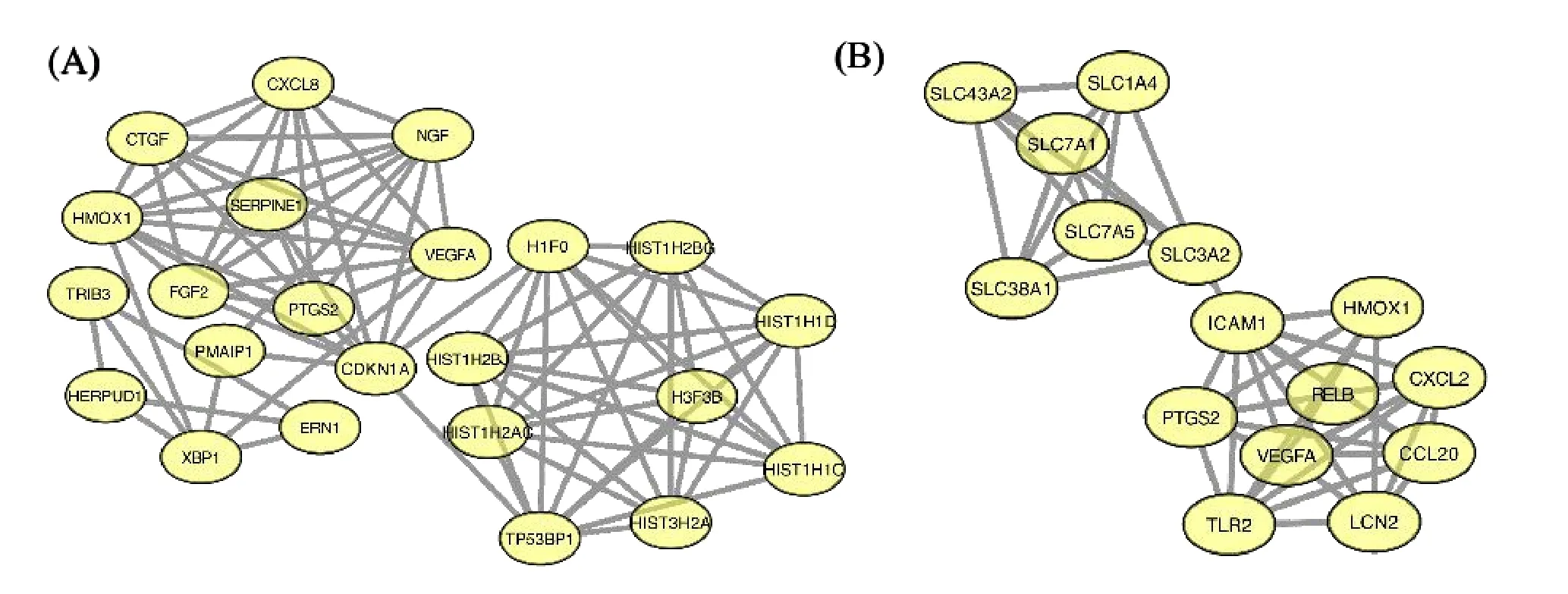
Figure 2 PPI networks of the DEGs in CCBS or HABS compared with NBS analyzed by STRING and Cytoscape softwares.(A)The most significant module was obtained from PPI network of the DEGs in CCBS.(B)The most significant module was obtained from PPI network of the DEGs in HABS.BSS,blood stasis syndrome;CCBS,cold-coagulation BBS;HABS,heat-accumulation BBS;NBS,non-BBS;DEGs,differentially expressed genes;PPI,protein-protein interactions.
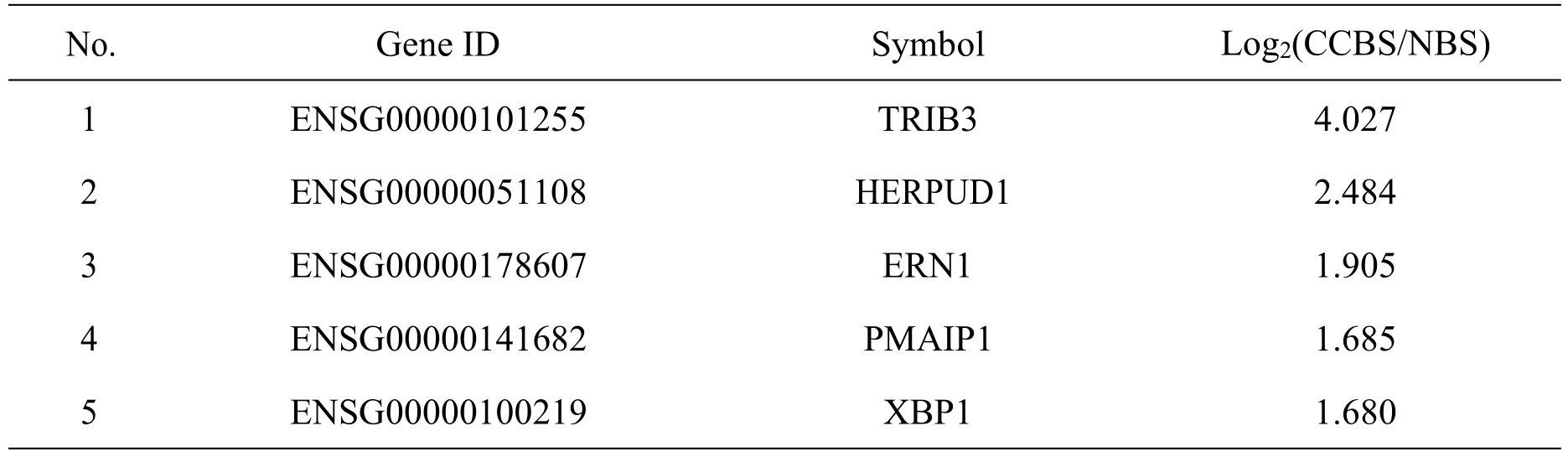
Table 3 High throughput expression results of the potential key genes in CCBS
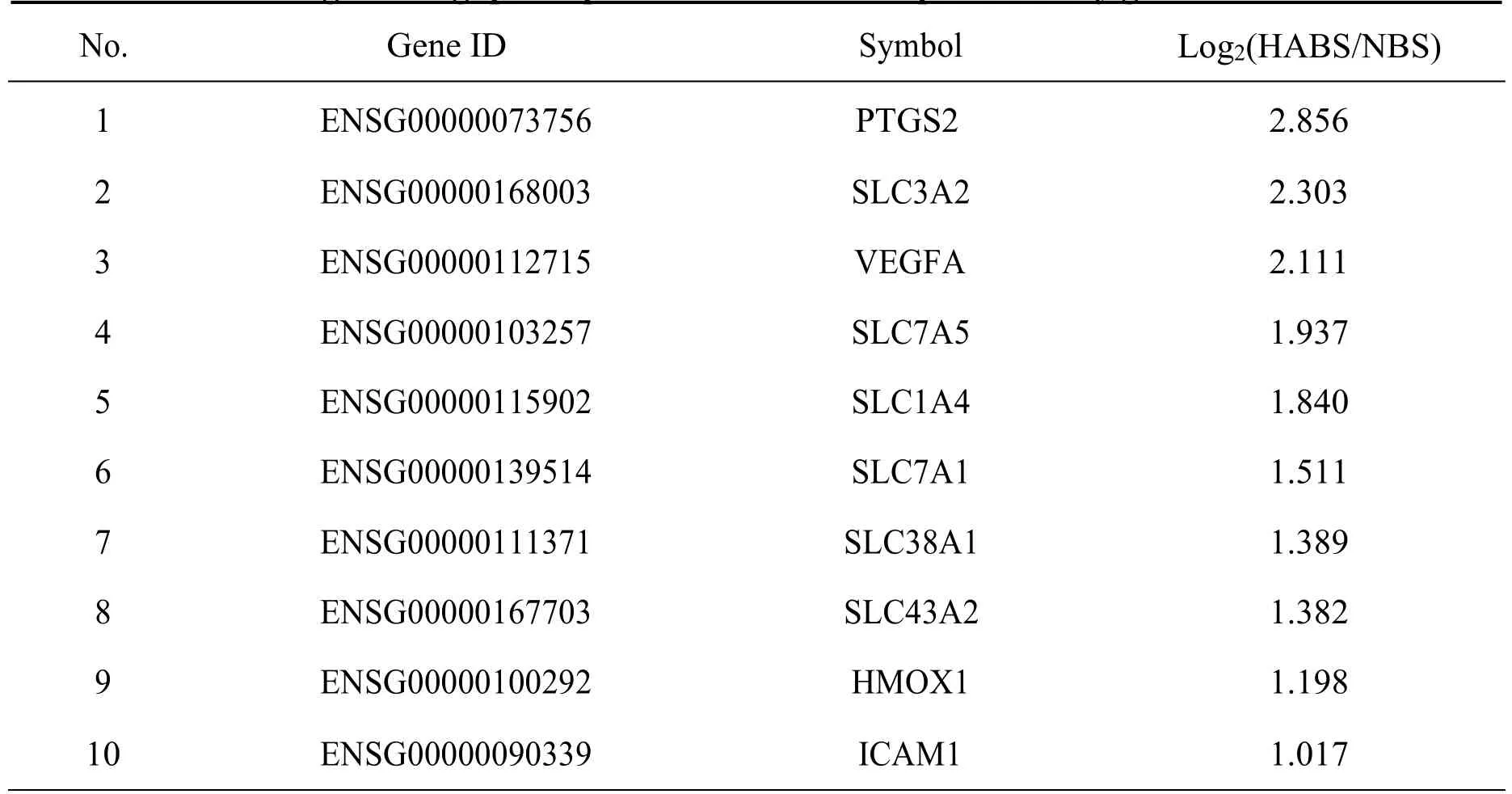
Table 4 High throughput expression results of the potential key genes in HABS
Discussion
BSS is a common traditional Chinese medicine syndrome,which is closely associated with endothelial cell dysfunction in many diseases,such as hypertension,coronary heart disease and diabetes mellitus,etc.In this study,we established CCBS,HABS and NBS endothelial cell models of hypertension.The DEGs of CCBS or HABS were obtained compared with NBS.Bioinformatics analyses of the DEGs were conducted,including GO,pathway,PPI networks,etc.Finally,5 genes were selected as the candidate genes of CCBS in hypertension,including TRIB3,HERPUD1,ERN1,PMAIP1 and XBP1.And 10 genes were selected as the candidate genes of HABS in hypertension,including PTGS2,SLC3A2,VEGFA,SLC7A5,SLC1A4,SLC7A1,SLC38A1,SLC43A2,HMOX1 and ICAM1.
In the study,368 DEGs were found to be associated with CCBS,and GO analyses showed that they were mainly enriched in intrinsic apoptotic signaling pathway in response to endoplasmic reticulum stress,which were proved to be closely related to endothelial dysfunction [15].The same results were found by pathway analyses of the DEGs of CCBS,which were mainly enriched in HIF-1 signaling pathway.It proved that DPP-4 inhibitors protect rat brain microvascular endothelial cells under hypoxic/high-glucose conditions potentially through the HIF-1/VEGF pathway[16].In addition,5 genes were selected as the candidate genes of CCBS in hypertension,including TRIB3,HERPUD1,ERN1,PMAIP1 and XBP1.It has proven that most of them were associated with endothelial function or hypertension.For example,TRIB3 mediating signaling pathways are closely related to blood pressure regulation [17].It was proved that miR-384 could negatively regulate HERPUD1 and protect human umbilical vein endothelial cells against angiotensin II-induced apoptosis [18].ERN1 and XBP1 and proved to be closely associated with hypoxia-induced pulmonary arterial hypertension [19].PMAIP1 was one of the molecular biomarkers of associated with hypertension or endothelial function.
There were 271 DEGs were found to be associated with HABS,and GO analyses showed that they were mainly enriched in cellular response to hypoxia,which were proved to be associated with pulmonary hypertension [20].KEGG pathway analyses showed that they were active in TNF signaling pathway,which were proved to be closely related to pulmonary endothelial injury [21].The 10 genes were chosen as the potential key genes of HABS in hypertension,including PTGS2,SLC3A2,VEGFA,SLC7A5,SLC1A4,SLC7A1,SLC38A1,SLC43A2,HMOX1 and ICAM1.PTGS2 was found to be associated with preecampsia and inflammation in human umbilical vein endothelial cells [22].SLC3A2 in human umbilical vein endothelial cells was associated with the inhibition of no synthesis in medium containing 1.5 microM L-arginine[23].VEGFA was closely related to endothelial function in cardiovascular diseases [24].SLC7A5 was associated with microvascular rarefaction/ischemia in pulmonary artery hypertension[25].Maternal undernutrition during pregnancy may lead to fetal intrauterine growth restriction and predisposes to adult risk of hypertension,which might be related to the downregulation SLC38A1[26].It was proved that a functionally active polymorphism in the 3'UTR of SLC7A1 may account for the altered endothelial function in essential hypertension [27].HMOX1 could suppress a pro-inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension [28].ICAM1 has long been used as a inflammatory mediator and marker of endothelial function,and be very closely related to hypertension[29,30].No evidence has been shown that SLC1A4 and SLC43A2 were associated with hypertension or endothelial function.
In short,the mechanism of BSS is very complex and involves many pathways.The main mechanism may be closely associated with activation of cellular endoplasmic reticulum stress,hypoxia inducible factor or tumor necrosis factor,etc.And the candidate genes can be used as biomarkers or drug targets for CCBS or HABS.This study took a new approach to select candidate genes for CCBS or HABS associated with hypertension by RNA sequencing and bioinformatics analyses.However,there are some limitations in this study,for example,some candidate genes were not confirmed the validity.Therefore,the role of the candidate genes should be identified in future studies.
杂志排行

Clinical Research Communications的其它文章
- The prognostic value of baseline circulating tumor cells in patients with metastatic breast cancer:a meta-analysis
- Meta-analysis of the clinical efficacy and safety of Urinary Kallidinogenase in the treatment of acute cerebral infarction
- Acute uterine inversion at cesarean section:an emergency condition in obstetrics
- Research on the aetiology and treatment of premature ovarian insufficiency