在中国使用左乙拉西坦和卡马西平单药治疗部分性癫痫发作的比较:一项开放性、非劣效性试验的主要结果
2020-11-23廖卫平周东ToruOsakabeChristianLoesch杜新鲁FrankTennigkeit王学峰
廖卫平,周东,Toru Osakabe,Christian Loesch,杜新鲁,Frank Tennigkeit,王学峰
在中国,已批准将左乙拉西坦作为部分性(局灶性起源)癫痫发作的添加治疗,片剂和静脉注射剂用于≥4岁的患者、口服液用于≥1个月的患者,还可作为部分性癫痫发作的单药治疗用于≥4岁的患者(片剂),作为全身强直-阵挛发作的添加治疗用于≥16岁的患者(片剂)。作为单药治疗用于体质量≥50 kg的成人(年满18岁)和青少年(12~17岁)时,建议最大剂量为3 000 mg/d。
在中国,卡马西平(CBZ)是最常用的抗癫痫药物(AED) 之一[1-2],适用于部分性癫痫发作以及原发性和继发性全身强直-阵挛发作。对于成人,通常在每天2~3次,每次400 mg的剂量下,治疗效果最佳。
在欧洲和南非开展的一项双盲、Ⅲ期试验(试验 N01061;NCT00150735)[3]表明,对于新诊断的部分性癫痫发作或全身强直-阵挛发作的患者,左乙拉西坦(1 000~3 000 mg/d)的疗效不低于 CBZ 控释剂(CBZ-CR,400~1 200 mg/d)。已经证实,对有部分性癫痫发作的中国患者,使用左乙拉西坦3 000 mg/d作为添加治疗,结果有效(试验 N01102;NCT00152373)[4],左乙拉西坦在中国健康志愿者体内的药代动力学(以下简称“药代”)特征与高加索白人的药代特征相近[5]。在日本人中也证实,左乙拉西坦单药治疗(1 000~2 000 mg/d)可有效治疗部分性癫痫发作(试验 N01375;NCT01506882)[6]。
根据美国处方信息,对于≥16岁的部分性癫痫发作患者,左乙拉西坦作为添加治疗的建议最低剂量是1 000 mg/d[7]。对于成人和>12岁的儿童,CBZ作为抗癫痫药物的建议最低初始剂量为400 mg/d[8]。本文所述试验的目的是比较左乙拉西坦(1 000 mg/d)和速释CBZ(CBZ-IR)(400 mg/d)单药治疗对新诊断或最近诊断为部分性癫痫发作的中国成人和青年患者(≥16 岁)的疗效,主要分析如下。
1 试验方法
1.1 试验设计 本试验是2013年9月至2015年9月在中国的28个研究中心开展的一项Ⅲ期、多中心、随机分配、开放性、平行组、阳性对照试验(N01364;NCT01954121)。此试验的主要目的是证明,在作为单药治疗至少使用6个月的情况下,左乙拉西坦(1 000 mg/d)的疗效不低于CBZ-IR(400 mg/d)。符合资格的患者根据预定的随机分配和预设试验流程,通过交互式语音应答系统或交互式网络应答系统,按1∶1的比例随机分配到左乙拉西坦(1 000 mg/d)组或CBZ-IR(400 mg/d)组。随机分配入组的患者再按照首次访视前3个月内的癫痫发作次数(≤2次癫痫发作或>2次癫痫发作)来分层。此试验包括最多1周筛选、2周上调剂量、1周稳定、最长26周评估和最多3周下调剂量(图1)。患者在上调剂量期开始接受试验药物,剂量是随机分配的目标剂量的一半,在上调剂量期结束时开始接受完整的目标剂量。患者如果没有达到或不耐受目标剂量,在26周评估期内出现癫痫发作,或者出现新的癫痫发作类型,则退出试验。
在评估期内没有癫痫发作的患者或者出现癫痫发作且需要进一步调整剂量的患者,有机会通过指定患者药物使用计划 (NPP) 继续接受随机分配给他们的试验药物。使用左乙拉西坦的患者,其剂量可上调至3 000 mg/d,且可以一直使用此药物,直到2017年7月UCB Pharma停止了该NPP计划。使用CBZ-IR的患者,其剂量可上调至1 600 mg/d,且可通过NPP继续使用此药达6个月,此后可由研究者斟酌决定,让其转用市售CBZ-IR处方药。
除了NPP以外,此试验依照《药物临床试验质量管理规范》、《赫尔辛基宣言》,以及当地法律开展。研究方案、修订和患者知情同意书由全国性、地区性或独立伦理委员会或机构审查委员会审查。所有患者(或其父母/法定监护人)就参加试验给予了书面知情同意。
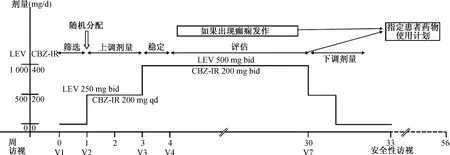
图1 试验设计。bid:每日2次;LEV:左乙拉西坦;qd:每日1次;V:访视
1.2 对象选择标准 符合资格的患者为中国人、≥16岁,新诊断或最近诊断为癫痫,曾有过非诱发性部分性癫痫发作[简单部分性发作(IA)、复杂部分性发作(IB)、部分性发作继发全身强直阵挛性发作(IC)]且有明显的局灶性起源。患者在随机分配前的1年内,必须报告过≥2次非诱发性癫痫发作(中间至少相隔48 h),其中≥1次非诱发性癫痫发作在随机分配前的3个月内发生。对于简单部分性发作,只计运动型发作的次数。排除标准:存在人白细胞抗原等位基因(HLA-B)*1502;曾有或目前有非部分性癫痫发作;仅出现过IA类非运动性癫痫发作;曾有仅丛集性癫痫发作;曾有特发性全身性癫痫指征;目前或曾经诊断为假性癫痫发作、转换性障碍,或其他可能与癫痫发作混淆的非癫痫发作事件;曾有癫痫持续状态;过去6个月内使用一种AED治疗癫痫(不包括≤2周的急性治疗);曾使用左乙拉西坦或CBZ治疗;过去6个月内使用传统中药治疗癫痫。
1.3 试验结果评估方法 主要疗效结果是达到6个月无癫痫发作(即整个26周评估期内无癫痫发作)的患者比例。次要疗效结果:在上调剂量期、稳定期和评估期这30周期间的患者保留率;评估期内首次癫痫发作的时间;从使用第一剂试验药物开始,经过上调剂量期和稳定期,一直到评估期的这段时间内,首次癫痫发作的时间;在评估期内,因不良事件(AE)或缺乏疗效导致首次癫痫发作或中止试验的时间。安全性和耐受性结果:治疗中出现的AE(TEAE)、TEAE 引起的中止试验、严重AE(SAE)、死亡和血液事件。
采用事后分析确定左乙拉西坦和CBZ-IR组中止试验的患者中进入NPP的患者比例,同样确定完成试验的患者中进入NPP的患者比例。
1.4 统计学方法 安全集(SS)包括至少使用过一剂试验药物的所有随机分配的患者。全分析集(FAS)包括SS中在评估期有癫痫发作日志数据的所有患者。符合方案集(PPS)包括FAS中进入评估期、且没有被视为会影响疗效判读的重大方案偏离的所有患者。原假设是左乙拉西坦1 000 mg/d的疗效低于CBZ-IR 400 mg/d。
如果在评估期的连续6个月内,没有癫痫发作记录;患者在评估期内没有提前中止试验,且没有漏记癫痫发作;而且评估期的总持续时间至少为182 d(26周),则认为患者的主要疗效指标为无癫痫发作。采用 Logistic 回归模型并考虑首次访视前3个月内的癫痫发作次数分类(≤2次癫痫发作和>2次癫痫发作),对主要疗效指标进行了分析。通过该模型预测的指标用于计算6个月无癫痫发作的患者比例的校正后绝对差值(左乙拉西坦-CBZ-IR),以及该差值的双侧 95%CI。如果CI下限高于-0.2,即认为左乙拉西坦的疗效不低于CBZ-IR,则原假设不成立。如果除了原假设不成立以外,CBZ-IR和左乙拉西坦的6个月无癫痫发作的预测比例>30%,则认为此试验的检验结果为阳性。
样本量根据一项相应的非劣效性检验来确定,该检验的把握度为90%,单侧显著性水平为2.5%,使用未校正差值和正态近似法[9],(绝对)非劣效性界值为 20%,且假设左乙拉西坦组的6个月无癫痫发作率为59%,CBZ-IR组为62%[3]。由此得出,PPS需要的可评估样本量为每组174例患者。考虑到脱落和因违背方案而被排除的患者,将此样本量增加了 25%[3],因而每组为218例患者,即436例患者被随机分配。使用比例风险模型分析不良事件的发生时间指标,该模型以治疗为因素,以首次访视前3个月内的癫痫发作次数分类(≤2次癫痫发作和>2次癫痫发作)为分层指标。从该模型中得出左乙拉西坦与CBZ-IR之间的风险比(HR)以及相应的95%CI。
2 结 果
2.1 入选患者分布和基线人口统计资料 在436例随机分配的患者中,433例纳入SS,374例纳入FAS,357例纳入PPS(图2)。

图 2 入选患者分布图。开始试验表示随机分配的患者人数;稳定期包含在上调剂量期内;只有在患者使用试验药物时,才存在上调剂量。a:这些比例的分母是随机分配集中接受左乙拉西坦的患者;b:这些比例的分母是随机分配集中接受 CBZ-IR 的患者
总体而言,436例患者中,有218例(50.0%) 完成了试验,其中左乙拉西坦组的220例患者中有93例(42.3%),CBZ-IR组的216例患者中有125例(57.9%);436例患者中,有218例(50.0%)中止试验,其中左乙拉西坦组的220例患者中有127例(57.7%),CBZ-IR组的216例患者中有91例(42.1%)。中止试验的最常见原因是缺乏疗效[左乙拉西坦组的220例患者中有94例(42.7%);CBZ-IR组的216例患者中有41例(19.0%)]和 AE[左乙拉西坦组的220例患者中有7 例(3.2%);CBZ-IR 组的216例患者中有 26例(12.0%)]。在中止试验的127例接受左乙拉西坦治疗的患者中,86例(67.7%)进入NPP;在中止试验的91例接受CBZ-IR治疗的患者中,35例(38.5%)进入NPP(χ2检验P<0.0001)。在完成试验的患者中,左乙拉西坦组的全部93例(100%)患者,以及CBZ-IR组125例患者中的123例(98.4%)进入了NPP。
两组的人口统计资料特征(表 1)大多相似,不过CBZ-IR组中患者的癫痫平均开始年龄略低于左乙拉西坦组患者(分别为32.7岁和37.7岁)。总体而言,2.5%的患者曾使用AED,最常见的是丙戊酸和地西泮。所有患者中6.2%曾使用过传统中药,最常见的是一种“草本制剂”[左乙拉西坦组中14例(6.4%),CBZ-IR组中10例(4.7%)]。左乙拉西坦组中有2例患者曾使用过银杏叶提取物,该组另外各有1例患者使用过苦橙和高丽参。

表1 患者基线人口统计资料和特征 (SS)特征(SS)左乙拉西坦组(n=218)CBZ-IR组(n=215) 合计(n=433)年龄[(均值±标准差),岁]37.8±16.233.3±14.335.6±15.4 ≤18岁(例,%)20(9.2)22(10.2)42(9.7) 18~65岁(例,%)184(84.4)186(86.5)370(85.5) ≥65岁(例,%)14(6.4)7(3.3)21(4.8)男性(例,%)112(51.4)121(56.3)233(53.8)体质量指数(BMI)[(均值±标准差),kg/m2]22.70±3.2222.35±3.2222.53±3.22进入试验时的癫痫持续时间[中位值(范围),月]0.0(0~171)0.0(0~306)0.0(0~306)癫痫开始的年龄[(均值±标准差),岁]37.7±16.532.7±14.935.2±15.9任何合并症(例,%)74(33.9)66(30.7)140(32.3)在整体中≥2%的患者存在的共患病(例,%)a 高血压25(11.5)10(4.7)35(8.1) 高血脂4(1.8)6(2.8)10(2.3)上一年痫性发作次数[中位值(范围)]5.0(2~1450)b4.0(2~752)b4.0(2~1450)c过去3个月痫性发作次数[中位值(范围)]2.5(1~510)d3.0(1~315)d3.0(1~510)e曾使用的 AED(例,%) 所有9(4.1)2(0.9)11(2.5) 丙戊酸f4(1.8)2(0.9)6(1.4) 地西泮3(1.4)03(0.7) 苯妥英1(0.5)1(0.5)2(0.5) 奥卡西平1(0.5)01(0.2) 苯巴比妥1(0.5)01(0.2) 注:a国际医学用语词典(MedDRA)第18.0版首选术语;bn=201;cn=402;dn=208;en=416;f使用的丙戊酸包括丙戊酸钠(左乙拉西坦组3例,CBZ-IR组2例)和丙戊酸(左乙拉西坦组1例)
2.2 疗效 在PPS集中,接受左乙拉西坦治疗的186例患者中有88例(47.3%)达到6个月无癫痫发作,接受CBZ-IR治疗的171例患者中有117例(68.4%)达到6个月无癫痫发作,校正后绝对差值为-22.9%(95%CI:-33.1%,-12.6%),此CI的下限低于非劣效性界值-20%(表 2)。FAS集的结果与PPS集相近(校正后绝对差值为-21.9%;95%CI:-32.0%,-11.9%)(表2)。

表2 患者疗效分析(PPS、FAS)项目PPS左乙拉西坦组(n=186)CBZ-IR组(n=171)FAS左乙拉西坦组(n=193)CBZ-IR组(n=181)6个月无癫痫发作的患者数(例,%)88(47.3)117(68.4)90(46.6)121(66.9)6个月无癫痫发作率的校正后绝对差值(左乙拉西坦-CBZ-IR)(95%CI,%)a-22.9(-33.1, -12.6)-21.9(-32.0, -11.9)非劣效性界值(%)-20-20 注:a治疗组的无癫痫发作比例校正后绝对差值,根据治疗组的无癫痫发作患者的校正比例得出。校正比例根据无癫痫发作 Logistic回归模型得出,该模型使用治疗和首次访视前3个月内的癫痫发作次数分类(≤2次痫性发作和>2次痫性发作)作为协变量
在PPS集,左乙拉西坦组在30周期间的患者保留率[48.4%(186例患者中有90例)]低于CBZ-IR组[70.2%(171例患者中有120例)]。在评估期内,使用左乙拉西坦的186例患者中有87例(46.8%)出现癫痫发作、因故中止试验或漏记癫痫发作,而使用CBZ-IR的171例患者中只有39例(22.8%);在左乙拉西坦与CBZ-IR的HR方面,CBZ-IR更优(HR=2.686;95%CI:1.838,3.927)。同样,在上调剂量期、稳定期和评估期,左乙拉西坦组的186例患者中有97例(52.2%)出现癫痫发作、因故中止试验或漏记癫痫发作,而CBZ-IR组的171例患者中只有57例(33.3%);左乙拉西坦与CBZ-IR的HR为1.881(95%CI:1.353,2.614)。在评估期内,左乙拉西坦组的186例患者中有88例(47.3%)因AE或缺乏疗效导致癫痫发作或中止试验,CBZ-IR组的171例患者中有45例(26.3%);在左乙拉西坦与CBZ-IR的HR方面CBZ-IR更优(HR=2.338;95%CI:1.629,3.356)。
2.3 安全性和耐受性
2.3.1 使用试验药物 总体而言,左乙拉西坦组的中位治疗持续时间为134.5 d(范围:1~226 d);CBZ-IR 组为203.0 d(范围:1~241 d)(SS)。在评估期内,左乙拉西坦组的中位药物剂量为1 000.0 mg/d(范围:750.0~1 187.5 mg/d),CBZ-IR 组为398.9 mg/d(范围:325.8~402.2 mg/d)。
2.3.2 治疗中出现的AE 左乙拉西坦组中报告TEAE的患者略少于CBZ-IR组(分别为61.9%和67.9%;SS)。在左乙拉西坦组,因TEAE而中止试验、因TEAE永久停用试验药物,以及需要调整剂量的TEAE数量均少于CBZ-IR组(表 3)。
左乙拉西坦组最常见的TEAE(≥7%)是鼻咽炎(18.3%)、头晕(15.1%)、嗜睡(9.2%) 和头痛(8.7%)。CBZ-IR组最常见的TEAE(≥7%)是鼻咽炎(14.9%)、头晕(8.4%)、上呼吸道感染和头痛(各 7.4%)。两个治疗组中,最常见的TEAE的严重程度均为轻度至中度。
试验期间,有1例被随机分配接受左乙拉西坦的患者死亡。该患者发生了癫痫 TEAE,此试验的研究者认为与试验药物无关。在左乙拉西坦组,γ-谷氨酰转移酶升高、WBC降低和肝功能检查异常的数量(分别为 0.9%、0.5% 和 0%)均低于CBZ-IR组(分别为5.1%、5.1%和4.2%)。
3 讨 论
此试验比较了左乙拉西坦(1 000 mg/d)和 CBZ-IR(400 mg/d)单药治疗对新诊断或最近诊断为部分性癫痫发作的中国成人和青年患者(≥16岁)的疗效。原假设是左乙拉西坦1 000 mg/d的疗效低于CBZ-IR 400 mg/d;然而无法拒绝无效假设,所以此试验的主要目的没有达到。
以往在欧洲和南非人中开展的一项Ⅲ期、随机分配、双盲、阳性对照、单药治疗试验 (N01061) 证明,对于新诊断为癫痫的患者,使用左乙拉西坦(1 000~3 000 mg/d)作为初始单药治疗的疗效不低于CBZ-CR(400~1 200 mg/d)[3]。在该实验中,使用最终评估的剂量[中位剂量:左乙拉西坦1 000 mg/d和CBZ-CR 400 mg/d(UCB Pharma 档案数据)]下的6个月无癫痫发作数据,评估了非劣效性。鉴于左乙拉西坦在中国人和高加索白人体内的药代特征相近[5],预计在中国患者人群中,左乙拉西坦的疗效也不低于CBZ-CR。本试验是根据 N01061 试验中报告的左乙拉西坦1 000 mg/d和CBZ-CR 400 mg/d的6个月无癫痫发作率来规划的(分别为59%和62%)[3]。

表3 治疗中出现的AE发生率(SS)(例,%)指标左乙拉西坦组(n=218)CBZ-IR组(n=215)任何TEAE135(61.9) 146(67.9) 严重TEAE9(4.1) 11(5.1) 因AE而导致中止试验12(5.5)29(13.5)因TEAE永久停用试验药物12(5.5) 29(13.5) 需要调整剂量的TEAEa22(10.1) 43(20.0) ADRb61(28.0) 80(37.2) 严重ADRb04(1.9)重度TEAE10(4.6) 12(5.6) 所有死亡1(0.5) 0任何一个治疗组中≥5%的患者报告的TEAEc,d 鼻咽炎40(18.3)32(14.9) 头晕33(15.1)18(8.4) 嗜睡20(9.2)7(3.3) 头痛19(8.7)16(7.4) 上呼吸道感染12(5.5)16(7.4) 尿路感染11(5.0)5(2.3) 血胆固醇升高6(2.8)11(5.1) 皮疹3(1.4)11(5.1) γ-谷氨酰转移酶升高2(0.9)11(5.1) WBC降低1(0.5)11(5.1)任何一个治疗组中导致≥1%的患者中止试验的TEAc,d 癫痫持续状态3(1.4)0 癫痫发作2(0.9)3(1.4) 皮疹07(3.3) 超敏反应03(1.4)任何一个治疗组中≥2%的患者报告的ADRb,c,d 头晕21(9.6)13(6.0) 嗜睡20(9.2)6(2.8) 头痛4(1.8)7(3.3) 皮疹2(0.9)10(4.7) γ-谷氨酰转移酶升高2(0.9)8(3.7) WBC降低1(0.5)8(3.7) 肝功检查异常08(3.7) 注:a需要调整剂量的TEAE是指导致撤药、暂停给药、增加剂量或不适用的TEAE;bADR是指关系为“相关”或缺失的TE-AE;cMedDRA第18.0版首选术语;dTEAE按左乙拉西坦组的发生率来排序
虽然在本试验中,左乙拉西坦的疗效低于预期的原因尚不明确,但未能证明其疗效不低于CBZ-CR,可能是由于本试验与N01061在设计上的根本差异造成的。N01061允许患者在评估期内使用当前AED剂量出现癫痫发作的情况下,将AED剂量最多提高两倍(左乙拉西坦最高剂量为3 000 mg/d,CBZ-CR最高剂量为1 200 mg/d)。然而在本试验中,仅允许使用一种低剂量的左乙拉西坦和CBZ-IR。按照研究方案,出现癫痫发作且需要进一步调整剂量的患者退出试验;但是,他们有机会通过NPP继续以更高的剂量使用随机分配给他们的试验药物进行治疗。左乙拉西坦组患者因缺乏疗效而中止试验的比例高于CBZ-IR组(分别为42.7%与19.0%),但因AE而中止试验的比例低于CBZ-IR组(分别为3.2%和12.0%)。由于上述试验设计特点,有理由推断会产生CBZ-IR更优效的结果偏差。此外,N01061为双盲试验,而本试验由于没有盲法对照药,所以是开放性试验。开放性设计可能会导致出现CBZ-IR患者保留率更高的偏差。由于研究者更熟悉标准治疗(即CBZ-IR),所以他们可能更倾向于让患者使用这种治疗的时间更久一些。另一方面,研究者对试验性治疗(即左乙拉西坦)的经验较少,可能会更快地中止治疗。
左乙拉西坦的耐受性良好,试验证明其安全性和耐受性情况优于CBZ-IR。在左乙拉西坦组,因TEAE而中止试验、因TEAE而停用试验药物,以及需要调整剂量的TEAE发生数量均少于CBZ-IR 组。这是排除了HLA-B*1502等位基因检测阳性的患者之后的试验结果,这可能导致CBZ-IR组更优效的结果偏差。对于中国裔患者,HLA-B*1502与严重、有时甚至危及生命的CBZ皮肤超敏反应风险密切相关,在香港、泰国、马来西亚和菲律宾部分地区的发病率超过15%,在台湾约为10%,华北约为4%,在南亚平均为2%~4%,日本和韩国为<1%[8]。
本试验中,使用左乙拉西坦1 000 mg/d的无癫痫发作率(47.3%)与一项对日本患者使用相同剂量的左乙拉西坦开展的试验中所报告的无癫痫发作率相近[57例患者中有28例(49.1%);UCB Pharma档案数据]。在中国开展的一项对比六种AED单药治疗有效性的大规模回顾性研究(n=789)证明,在首次癫痫发作时间、减少癫痫发作频率、缓解率以及中止试验的时间方面,左乙拉西坦与CBZ和奥卡西平的效果相近,但是在六种药物中的AE发生率最低,且该发生率显著低于托吡酯、丙戊酸或CBZ[10]。CBZ和拉莫三嗪最常引起因AE导致治疗失败;此外,据报告,使用CBZ还会出现肝功能异常和WBC下降,导致一些患者中止试验[10]。因而,笔者推荐使用左乙拉西坦来治疗部分性癫痫发作患者,尤其是有系统性疾病的患者[10]。对于有共患病和使用其他药物的患者,选择一种不会发生药物间相互作用的AED也很重要[11]。此外,初始药物也很重要,因为在多种AED治疗失败后,减少癫痫发作的可能性会更低[12]。
根据国际抗癫痫联盟2013年的总结,左乙拉西坦作为部分性癫痫发作成人患者的初始单药治疗,其疗效/有效性为A级[13]。此外,在中国和日本,左乙拉西坦是推荐使用的部分性癫痫发作一线治疗[14-15]。本试验提供了对新诊断或最近诊断为部分性癫痫发作的中国患者,采用左乙拉西坦单药治疗与标准AED相比的疗效和安全性数据。在低剂量单药治疗下,无法确定左乙拉西坦的疗效不低于CBZ-IR。由于试验设计为开放性、固定剂量,且所评估的剂量水平低,所以疗效结果难以判读。需要上调剂量以控制癫痫发作的患者必须退出试验,但有机会通过NPP接受更高的剂量。尽管有这些局限,但观察到的6个月无癫痫发作率表明,左乙拉西坦可能是一种有效的单药治疗药物。左乙拉西坦的安全性结果与已知安全性特征一致,其安全性和耐受性情况优于CBZ-IR。
志谢:本试验由UCB Pharma资助。笔者感谢患者及其照顾者,感谢陈生弟教授(上海交通大学医学院附属瑞金医院)、邓艳春教授(空军军医大学西京医院)、洪震教授(复旦大学华山医院)、胡兴越教授(浙江大学医学院附属邵逸夫医院)、黄一宁教授(北京大学第一医院)、林宏教授(空军军医大学唐都医院)、刘建仁教授(上海交通大学医学院附属第九人民医院)、刘煜敏教授(武汉大学中南医院)、陆钦池教授(上海交通大学医学院附属仁济医院)、潘小平教授(广州市第一人民医院)、孙红斌教授(四川省人民医院)、孙美珍教授(山西医科大学第一医院)、谭兰教授(青岛市立医院)、万琪教授(江苏省人民医院)、王维平教授(河北医科大学第二医院)、王文敏教授(昆明医科大学第一附属医院)、王小姗教授(南京脑科医院)、王赞教授(吉林大学白求恩第一医院)、徐小林教授(天津环湖医院)、徐运教授(南京鼓楼医院)、张黎明教授(哈尔滨医科大学附属第一医院)、张志珺教授(东南大学附属中大医院)、赵合庆教授(苏州大学附属第二医院)、周列民教授(中山大学附属第一医院)、周晓红教授(广东省人民医院)以及他们的团队中为本试验作出贡献的所有人。笔者感谢专业医学撰稿人Kyoko Hirano(日本东京 UCB Pharma)和Barbara Pelgrims博士(比利时布鲁塞尔UCB Pharma)审核本稿并监督排版。Emily Chu 博士(英国伦敦Evidence Scientific Solutions)和专业医学撰稿人Richard Fay博士(美国宾州费城Evidence Scientific Solutions)协助编写,此项工作由UCB Pharma资助。Jessica Gamage(英国马格斯菲特UDG Healthcare plc旗下的一家阿什菲尔德公司QXV Communications)和Azita Tofighy(英国伦敦)提供初稿,此项工作由UCB Pharma资助。
披露/利益冲突:Toru Osakabe、Christian Loesch和Frank Tennigkeit是UCB Pharma的员工。Toru Osakabe获得UCB Pharma的员工股票。Frank Tennigkeit获得UCB Pharma的员工股票期权。杜新鲁在开展此试验时仍是UCB Pharma的员工。廖卫平、周东和王学峰申报没有利益冲突。
