血管麻痹综合征的潜在治疗靶点: 氧自由基
2020-11-17俞莹黄巧文翁险峰张良成彭勇刚
俞莹, 黄巧文, 翁险峰, 张良成, 彭勇刚
(1福建医科大学附属协和医院麻醉科, 福建 福州 350001; 2福建医科大学附属漳州市医院麻醉科, 福建 漳州 363000; 3美国佛罗里达大学Shands医院麻醉科, 美国 佛罗里达州 32610)
血管麻痹综合征(vasoplegic syndromes, VS)的理念由Gomes的研究团队在心脏手术中首次报道[1]。 VS的特征: 严重低血压, 正常或正常偏高的心输出量, 低全身血管阻力以及对血容量和升压药的需求增加。 据统计, 心脏移植术患者VS发生率为 30.8%, 接受体外循环冠状动脉搭桥术(coronary artery bypass graft, CABG)的患者中VS的发生率为6.9%, 非体外循环CABG的患者VS发生率为2.8%, 左心室辅助装置放置后VS发生率为29.0%, 而在烧伤、 创伤和胰腺炎中VS也有病例报道[2]。 研究显示[3], 重症监护病房(ICU)所有形式的休克中, 血管麻痹性休克可高达68.0%, 而脓毒症约占血管麻痹性休克的91.0%, 同时研究证实, 去甲肾上腺素难以纠正的VS, 死亡率更高。 一组针对1992例体外循环的心脏患者的数据分析显示, 405例VS患者(占总数20.3%)中, 中度和重度VS发生率分别为5.7%和1.5%[4]。 有研究显示, 所有心脏手术术后的患者, 其中25.0%的患者VS持续超过36~48 h[5]。 有研究表明, ICU患者的VS也与不良预后直接相关[3]。 同时, VS中大剂量血管收缩药物的治疗还可能导致上、 下肢远端缺血和肠系膜缺血。 上述VS的发生多伴有严重的组织缺氧、 全身炎症反应、 全身多器官功能衰竭。 因此, 需要积极管理VS, 以降低术后并发症和死亡率。
1 VS的定义
到目前为止对于VS尚无严格的定义, 有文献提议以下标准作为VS的定义:(1)低血压[平均动脉压(MAP)<50 mmHg]; (2)低外周循环阻力(systemic vascular resistance, SVR)[SVR<1600 dyn·seg/(cm5·m2)]; (3)正常或高心输出量[心指数>2.2 L/(min·m2)]; (4)对传统的儿茶酚胺治疗无效[去甲肾上腺素输注>0.15 μg/(kg·min)][2](图1)。

图1 VS定义及相关机制
2 VS的危险因素
VS的危险因素很多, 将其大致划分为术前危险因素和术中危险因素(表1)。

表1 VS的危险因素
a 手术与VS发生率对比: 换瓣术>CABG; 主动脉换瓣或修补 3.1.1 三磷酸腺苷(adenosine triphosphate, ATP)敏感型钾离子通道(KATP通道)功能紊乱 细胞内ATP水平降低、 酸中毒等可促进血管平滑肌KATP通道的开放, 并诱导了K+从细胞流出, 从而导致血管平滑肌细胞膜超极化。 这种超极化使电压门控的Ca2+通道开放, 阻止Ca2+进入细胞内(图1), 最终通过降低细胞中Ca2+水平诱发去甲肾上腺素难治性的血管扩张[7]。 3.1.2 血管加压素V1A受体下调 生理条件下, 血管加压素作用于V1A受体通过阻断ATP敏感性钾通道加速Ca2+进入细胞, 改善由血管扩张导致的低血压症状(图1)。 脓毒血症休克时, 血管加压素受体缺乏或血管加压素受体的基因表达下调是血管加压素对VS治疗效果不佳的原因[8]。 3.1.3 核因子-κB(nuclear factor-κB, NF-κB)活性增加 临床出现的脓毒症休克和实验诱发的脓毒症休克中, 均观察到了NF-κB活性的显著增加(图1)。 有研究通过注射NF-κB siRNA使特定的NF-κB(p105)基因沉默, 结果显示48 h后NF-κB(p105)mRNA较前降低(60±8)%, 而抑制NF-κB活性可以显著降低细胞因子的诱导, 并促进Ca2+内流[9]。 3.1.4 腺苷释放 腺苷是一种由上皮细胞和肌细胞释放的强大的内源性血管扩张剂, 其致血管扩张机制与减少钙离子内流密切相关[10](图1)。 在主动脉瓣置换术前、 术中、 术后, 都显示血浆腺苷水平明显升高, 而且腺苷水平的变化与临床血流动力学波动显著相关。 该研究中确诊的VS患者显著升高的腺苷水平, 均提示腺苷的释放可能与心脏手术期间发生的全身性炎症反应的VS有关[10]。 3.2.1 内皮型一氧化氮合酶(endothelial nitric oxide synthase, eNOS)激活 eNOS激活引起的血管中NO含量增加是目前被研究最多, 也最被认可的引起VS的机制之一(图1)。eNOS是一种在血管内皮细胞中表达的钙依赖性酶。 当血管内皮受到剪切应力如发生全身炎症反应综合征(SIR)时, 内毒素血症以及白介素-1(IL-1)、 IL-6和肿瘤坏死因子-α(TNF-α)等均可导致内皮细胞中的钙动员, 从而以钙调蛋白依赖性方式激活eNOS。 而eNOS是血管NO产生的关键酶, 在各种辅助因子[例如(烟酰胺腺嘌呤二核苷酸磷酸, nicotinamide adenine dinucleotide phosphate, NADPH)和四氢生物蝶呤]存在下, eNOS将其生理底物L-精氨酸转化为NO和L-瓜氨酸[11]。 NO通过使细胞内钙离子降低, 肌球蛋白去磷酸化导致VS。 同时, NO又与其目标酶可溶性鸟苷酸环化酶的血红素基团结合并反过来激活eNOS。 eNOS的生物学作用也包括血管舒张和新生血管生成。 此外, 炎性细胞因子和病原体相关分子模式(如脂多糖等)可激活钙依赖性诱导型一氧化氮合酶(inducible nitric oxide synthase, iNOS)的合成, 导致NO较基线水平增加2~3倍, 这也是休克尤其是脓毒血症型休克时出现血管功能障碍的主要驱动因素[12]。 健康的内皮是血管维持张力和完整性的基础, 健康的内皮功能丧失被称为“血管内皮功能障碍”, 在内皮功能障碍中, 血管收缩和舒张物质之间的平衡被破坏, 是导致内皮收缩/舒张功能显著降低的主要原因。 而以氧、 氮、 碳为主体的自由基为强氧化剂, 可无差别破坏包括DNA、 脂质、 蛋白质在内的许多大分子。 氧自由基不仅直接损伤内皮细胞, 破坏收缩和舒张功能并且增加了血管炎性反应, 促进血栓形成, 其机制可能与PI3K-AKT通路激活有关[16]。 综上所述, 氧自由基引起的细胞损伤和血管通透性增加是血管病理性扩张的基础。 VS诊断的重要条件是血管对外源性血管活性药的反应性降低。 随着研究成功使用ONOO-模拟出多种与休克相关的血管变化(内皮功能障碍, 血管反应性低下), 其在循环性休克相关的血管变化中的机制也逐渐被揭示。 而ONOO-导致血管活性药物“失效”的机制, 是通过抑制线粒体功能和/或ONOO-直接氧化血管活性药物如多巴胺等使后者失活并降低收缩反应[19]。 而另一种血管活性药物去甲肾上腺素在VS中“失效”则与ONOO-直接使去甲肾上腺素受体失活有关, 在不同的实验中都观察到ONOO-介导的α-肾上腺素受体功能的抑制[20]。 当前没有一个确切的药物可有效治疗VS。 VS血液动力学治疗的主要目标是恢复平均动脉压(MAP)、 心输出量和改善微循环。 仅用血管活性药物维持微循环的稳定相对单纯维持血液更为困难。 如前所述, VS期间的血管内皮收缩功能损伤和对血管活性药物的低反应是多因素共同造成的。 近年来, 随着氧自由基在VS的作用机制被揭示, 一些已在临床使用的药物在减少氧自由基方面的作用逐渐被揭示。 而另一些相应的药物也进入了研究阶段。 总之, 包括VS一线用药的亚甲蓝(methylene blue, MB)、 维生素B12、 维生素C等明确具有还原效果的药物可通过氧化还原反应修复受损的内皮细胞, 改善血管的收缩功能。 而包括SOD模拟物, 亚硝酸盐中和/分解物等不同类型的药物则通过减少过氧化阴离子、 亚硝酸盐等致力于预防氧自由基对血管内皮的直接攻击, 从而弥补一些一线用药效果不确切的缺点。 MB是VS的一线药物, MB作为公认还原剂, 可以通过多种途径在VS中发挥其重要的抗氧化作用, 包括产生阻断作用的氧化剂和增强抗氧化剂的防御能力[5]。 MB在水和有机溶剂中具有广泛溶解性以及较低的氧化还原电位, 这些性质使MB可以轻松穿透双层膜并到达不同的细胞区(如线粒体)并在其氧化形式MB和还原形式MBH2之间快速交换。 当进入线粒体时, MB防止了线粒体中有毒副产物氧化剂的电子泄漏[5]。 总之, 这些数据支持MB促进线粒体功能, 并减少氧自由基的产生。 Mehaffey 等[21]指出早期使用MB对心脏术后VS的治疗非常有效, 并且没有明显副作用。 而在脓毒症休克和外周血管阻力低的患者中, 有研究也推荐使用MB[5]。 目前临床最常用的MB剂量为大剂量静脉注射2 mg/kg的MB, 此后以0.5 mg/(kg·h)的剂量连续输注12 h以维持稳定的血浆药物浓度[21]。 维生素C目前也被用于临床治疗VS, 其机制是其作为抗氧化剂通过防止ROS引起的四氢生物叶酸的氧化和随后的NO升高对已经损伤的血管内皮起到了保护和修复作用。 同时, 防止超氧化物与儿茶酚胺的相互作用以及形成称为肾上腺素的化合物, 而有助于恢复eNOS, 而铬对血压没有影响[22]。 研究发现, 高剂量维生素C可抵消内皮功能障碍, 而维生素C作为儿茶酚胺内源性生物合成必不可少的辅助因子改善了去甲肾上腺素的血管反应[22]。 1期临床研究证实以目前常用的维生素C静脉注射[负荷剂量1000 mg, 此后以200 mg/(kg·d)的剂量维持]可改善脓毒症VS患者的预后[23]。 人体内发现的维生素B12有4种形式, 其中羟钴胺素被广泛应用于临床, 静脉注射羟钴胺素已被证实可使健康成人MAP即刻升高并且持续4 h。 其机制是羟钴胺素选择性抑制iNOS, 从而减少ONOO-的产生并最终抑制炎症反应。 有研究总结了脓毒血症所致VS小鼠模型和猪模型中, 使用羟钴胺素可明显改善VS的生存率[24]。 Shapeton等[25]总结了7个临床中心的报道发现在10~15 min内静脉输注5 g(1例患者15 min静脉输注10 g)羟钴胺素均可有效改善血流动力学, 降低血管活性药物使用量并最终改善VS。 FeTMPyP[N-甲基-4’-吡啶基-卟啉铁(Ⅲ)]、 FeTMPS [2, 4, 6-三甲基-3, 5-二磺酰基苯基-卟啉铁(Ⅲ)]、 FeTPPS[5, 10, 15, 20-四(4-磺酰基苯基)-卟啉铁], ww-85等是过氧亚硝酸盐分解的催化剂, 已被证实可在大鼠脓毒症休克中可显著减少脂质过氧化和上皮细胞氧化性损伤[30], 改善心、 肾功能并促进循环稳定, 最终改善脓毒症休克的生存率[31]。 过氧亚硝酸盐分解催化剂FP15在内毒素休克和微生物败血症的大鼠和小鼠模型。 减少肝损伤, 改善低血压, 提高生存率[32]。 而MwTBAP作为过氧亚硝酸盐还原剂, 在脓毒血症休克中改善外周微血管过度扩张, 改善脓毒性休克预后[33]。 以上药物并未真正用于经典的VS的临床治疗, 但基于VS的发病机制及上述药物的作用机制, 这些药物具有研发的潜力, 可能需要更多的研究和数据支持其用于VS的治疗。 随着VS在心血管等大手术及脓毒血症休克中被逐渐认识, 人们对VS的定义开始清晰。 尽管维持动脉压, 保证足够的心输出量, 改善微循环是每一个临床工作者治疗VS的宗旨, 但在去甲肾上腺素等血管活性药物治疗效果不佳的前提下, 对VS机制更深入的探讨显得愈发重要。 在此基础上, 氧自由基的异常增多和消除减少在一定程度上解释了血管活性药物反应不佳的病理性血管扩张的机制。 而使用针对氧自由基相关的药物, 如NG-单甲基-L-精氨酸、 MB、 维生素C、 羟钴胺素等已被证实可以降低氧自由基作用, 一些新的药物如M-40403等在动物实验上也证明了其疗效, 未来相信针对氧自由基的治疗会成为治疗VS的新靶点。3 VS的发病机制
3.1 细胞内钙离子水平的降低
3.2 间接降低细胞内钙离子浓度
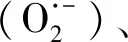
4 氧自由基在VS中的作用机制
4.2 ONOO-

5 针对氧自由基的治疗
5.1 MB
5.2 维生素C
5.3 维生素B12
5.4 NO抑制物
5.5 SOD模拟物

5.6 过氧亚硝酸盐还原剂或过氧亚硝酸盐分解催化剂
6 结语
