一个早发型家族性AD家系基因与脑葡萄糖代谢分析
2020-11-16张雅梦宋敬卉朱晓岩孙雪纯程雨菲马爱军
张雅梦 宋敬卉 朱晓岩 孙雪纯 程雨菲 马爱军

[摘要] 目的 分析一个早发型家族性阿尔茨海默病(AD)家系的大脑葡萄糖代谢特点并明确其基因诊断。方法 收集并分析该家系成员的临床资料,对该家系中现存活的4例病人进行18F-FDG PET脑显像,分析其大脑葡萄糖代谢特点,应用二代测序及Sanger测序方法检测早老素蛋白1(PSEN1)基因突变位点。结果 AD病人大脑葡萄糖代谢特点为双侧额叶、颞顶联合皮质区、颞叶外侧部及皮质下结构的代谢减低。Sanger测序发现此家系携带PSEN1基因突变(c.417G>c,p.M139I),为错义突变。结论 本研究在中国人群中首次报道了PSEN1基因p.M139I位点突变,扩大了PSEN1基因突变谱,该位点突变可能导致早发型家族性AD。
[关键词] 阿尔茨海默病;早老素1;突变;大脑;葡萄糖代谢障碍;氟脱氧葡萄糖F18Symbol`@@
[中圖分类号] R745.7;R394 [文献标志码] A [文章编号] 2096-5532(2020)06-0662-05
doi:10.11712/jms.2096-5532.2020.56.150 [开放科学(资源服务)标识码(OSID)]
[网络出版] https://kns.cnki.net/kcms/detail/37.1517.R.20200714.1202.002.html;
[ABSTRACT] Objective To investigate the features of cerebral glucose metabolism and genetic diagnosis of a family with early-onset familial Alzheimer disease (AD). Methods Clinical data were collected from the members of this family, and 18F-FDG PET brain imaging was performed for the four surviving patients in this family to analyze the features of cerebral glucose metabolism. Next-generation sequencing and Sanger sequencing were used to detect the mutation sites of the presenilin 1 (PSEN1) gene. Results The features of cerebral glucose metabolism were reductions in metabolism in the bilateral frontal lobes, the temporoparietal cortex, the lateral temporal lobe, and the subcortical structure in the patients with AD. Sanger sequencing showed that this family carried a missense mutation of the PSEN1 gene (c.417G>C, p.M139I). Conclusion This study reports the p.M139I mutation of the PSEN1 gene in the Chinese population for the first time, which expands the mutation spectrum of the PSEN1 gene, and mutation at this site may lead to early-onset familial AD.
[KEY WORDS] Alzheimer disease; presenilin-1; mutation; cerebrum; glucose metabolism disorders; fluorodeoxyglucose F18
阿尔茨海默病(AD)是老年痴呆最常见的类型,其特征是记忆和其他认知领域的进行性损伤[1]。以发病年龄65岁为界,可以分为早发型AD(EOAD)和晚发型AD(LOAD),其中EOAD是常染色体显性遗传病,占AD的2%~10%[2]。早老素蛋白1(PSEN1)[3]、早老素蛋白2(PSEN2)[4]、β淀粉样前体蛋白(APP)[5]基因的突变是EOAD的重要遗传致病因素,其中PSEN1是EOAD最常见的突变基因,突变频率为18%~50%[6]。本课题组既往报道了一个EOAD家系[7],其家庭成员在40岁左右出现以认知功能下降为特征的痴呆症,5代人中已有3代发病,最近家系中再次发现1例新发男性病例,发病者已达7例。该家系病人发病年龄轻,病情进展迅速,病程仅5~7年,国内少见类似报道。本研究对现存活的第3代的4例病人进行18F-FDG PET/CT脑显像检查,分析大脑葡萄糖代谢特点,并对家系成员进行基因测序,寻找致病基因。
1 资料和方法
1.1 一般资料
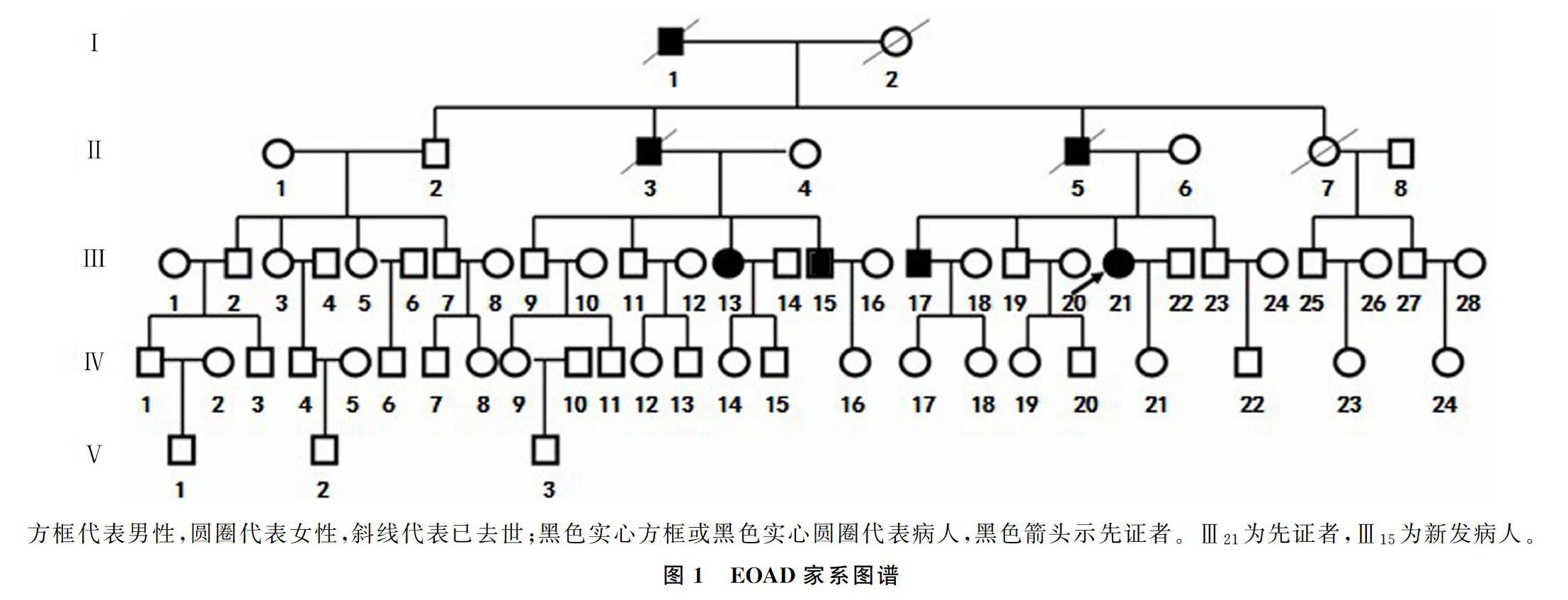
家系共65名成员,均为汉族,男性33人,女性32人。其中Ⅲ21为先证者,女,于36岁出现近事记忆减退,并出现暴躁、固执易怒等人格方面的障碍。先证者的父亲(Ⅱ5),于42岁出现遗忘、反应迟钝,并逐渐出现人格改变、定向障碍,生活不能自理,于49岁死亡。先证者的祖父(Ⅰ1),45岁发病,表现为记忆力下降、反应迟钝、人格改变等,50岁左右死亡,死因不明。先证者的伯父(Ⅱ3),43岁发病,症状类似,50岁时死亡。先证者的兄长(Ⅲ17)和堂姐(Ⅲ13)分别于42岁和43岁出现健忘、计算力下降、反应迟钝。新发病人(Ⅲ15)为先证者的堂弟(40岁),于2016年出现遗忘,以近事遗忘为主,伴计算能力下降,并逐渐出现反应迟钝、人格改变、定向障碍等;查体:发育正常,营养良好,神志清楚,表情淡漠,言语流利,记忆力、计算力、定向力下降,理解力尚保留,脑神经检查无阳性体征,四肢肌力、肌张力正常,腱反射对称,感觉及共济运动正常,病理征未引出。家系图见图1。本研究所有受试者在神经心理学测试、PET检查及基因检测前均经过本人或者其法定代理人的知情同意。
1.2 研究方法
1.2.1 神经心理学检查 对新发病人Ⅲ15进行神经心理学检查,涉及记忆、语言、定向力、应用能力、注意力、知觉(视、听、感知)和执行能力等7个领域。①简易精神状态量表(MMSE)用于临床初筛,最高得分为30分,27~30分为正常,<27分为认知功能障碍。痴呆严重程度分级:轻度,MMSE≥21分;中度,MMSE 10~20分;重度,MMSE≤9分。②日常生活能力量表(ADL)用于评定被试者的日常生活能力,最高64分,总分16分为完全正常,>16分表明有不同程度的功能下降。③Haehinski缺血量表(HIS)仅用于血管性痴呆和老年性痴呆的鉴别诊断,满分18分,得分在4分以下者,属老年性痴呆;7分以上者,则属血管性痴呆。
1.2.2 PET/CT检查及图像处理和分析 PET/CT检查前行颅脑CT或MRI检查排除脑出血、肿瘤等脑器质性病变。PET扫描儀型号为GE Discovery LS PET/CT。受试者(Ⅲ13、Ⅲ15、Ⅲ17和Ⅲ21)检查前空腹至少6 h,避免高糖饮食,于肘静脉注射示踪剂18F-FDG,休息45 min后进行PET脑显像。以PET/CT中心的健康查体者作为健康对照。图像处理及分析采用视觉分析法和SPM分析法,在Mtalab平台上,应用SPM 10软件对图像进行预处理和统计学分析,根据糖代谢变化区域的Talariach坐标值确定各脑区。
1.2.3 基因组 DNA提取 采集包括先证者在内的现存活的4例病人及49例健康家系成员的外周血5 mL,置于EDTA抗凝管中,使用血液基因组DNA抽提试剂盒(北京天根科技有限公司)提取血液样本的基因组DNA。
1.2.4 PSEN1基因突变检测 在金域医学检验集团股份有限公司协助下对提取的血液样本基因组DNA进行痴呆相关基因(PSEN1、PSEN2、APP和APOE等)的检测。
1.2.5 Sanger测序验证 引物由生工生物工程技术(上海)股份有限公司设计与合成。PCR反应体系共20 μL。反应条件如下:95 ℃预变性5 min;95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸1 min,循环30次。反应完成后对所得PCR产物进行测序。应用Chromas软件分析图像,应用NCBI Blast对测序结果进行序列比对,并排除基因多态性现象。
2 结 果
2.1 神经心理学评估
新发病人Ⅲ15 MMSE评分18分,量表总分及各单项得分均有不同程度地降低;ADL评分28分,明显大于16分,表明日常生活自理能力明显下降;HIS评分0分,排除血管性痴呆。
2.2 18F-FDG PET/CT脑显像分析
视觉法分析显示,大脑皮质示踪剂呈局灶性不均匀分布,双侧颞顶联合皮质区、额叶及颞叶外侧部等脑区示踪剂摄取减少(图2)。SPM法分析显示,双侧颞顶联合皮质区、颞叶外侧部、额叶及皮质下结构代谢减低,以颞顶联合皮质区及额叶为著(图3)。
1~5列为正常对照:脑部各结构显示清晰,大脑皮质、皮质下结构及小脑示踪剂分布均匀、对称;6~10列为病人Ⅲ15:双侧额叶、颞顶联合皮质区及颞叶外侧部等脑区示踪剂摄取减少。
2.3 DNA测序
该家系Ⅲ21(先证者)、Ⅲ13(先证者堂姐)、Ⅲ17(先证者兄长)、Ⅲ15(先证者堂弟)均在PSEN1基因的第5外显子发现第417位核苷酸由鸟嘌呤(G)突变为胞嘧啶(C),导致第139号密码子编码的氨基酸由蛋氨酸(Met)突变为异亮氨酸(Ile),描述为p.M139I;APP、PSEN2及APOE基因无异常。家系内健康成员基因组DNA中均未发现PSEN1、APP、PSEN2和APOE基因突变。见图4。
3 讨 论
AD是一种与年龄有关的神经退行性疾病,主要表现为记忆力下降、认知障碍、视空间损害、执行功能障碍以及人格和行为改变等,其特征性病理改变为β淀粉样蛋白(Aβ)的沉积、神经纤维缠结、神经元变性和突触缺失等[8-9]。由于AD病人脑内神经元大量丢失和突触活性减低,神经元对能量需求减低,进而导致大脑的葡萄糖代谢减低,因此FDG PET脑显像能够在体外反映AD病人大脑不同部位对FDG的摄取,从而反映大脑葡萄糖代谢的特点[10]。AD病人18F-FDG PET脑显像以颞顶叶、后扣带回、楔前叶及额叶低代谢为主要特征[11]。本家系中病人18F-FDG PET脑显像结果显示,大脑双侧额叶、颞顶联合皮质区、颞叶外侧部及皮质下结构葡萄糖代谢减低,符合AD的大脑葡萄糖代谢特征。传统的影像学检查手段,如颅脑CT和MRI,主要用于排除颅内占位性病变、脑出血、脑梗死等疾病和发现AD的特异性影像学表现,如脑萎缩和脑室扩大等,而18F-FDG PET在临床症状和脑结构出现变化之前,即可发现AD脑内异常功能变化,提示18F-FDG PET检查有利于早期AD的鉴别诊断[12],可以为研究不同发病年龄AD病人的大脑葡萄糖代谢特征提供线索。
家族性AD(FAD)是指家族中有连续2代或者2代以上,至少2位一级亲属罹患AD,且发病年龄小于60岁。常染色体显性遗传的EOAD可由已知的PSEN1、PSEN2和APP基因突变所致,这3个基因中的AD连锁突变具有高度外显性(>85%),因此,这3个基因被认为是EOAD的“诊断生物标志物”[13]。但APP、PSEN1和PSEN2的致病性突变只能解释早发型FAD家系的一小部分,大量遗传原因不明的EOAD病人表明有其他的致病基因尚未被鉴定[14]。APOEε4等位基因的纯合性是LOAD的主要遗传危险因素,是显著增加EOAD风险的独立遗传因素[15]。但是,与APP、PSEN1和PSEN2的突变不同,APOEε4等位基因不被认为是导致EOAD的重要原因,而仅仅是一个危险因素[16]。全基因组测序和全外显子测序等二代测序技术还发现,TYROBP、NOTCH3和SORL1可能是引起EOAD的基因[17-18]。近20年以来,在AD病人中发现了超过200个PSEN1基因突变,其中大多数是致病性的。约70%的PSEN1基因突变发生在第5~8外显子[19]。本研究对一个AD家系进行常见致病基因筛查,发现PSEN1基因第5外显子第417位核苷酸由G突变为C(c.417G>C),致使PSEN1编码蛋白第139位蛋氨酸被异亮氨酸替代,即p.M139I。先前有研究描述了M139I基因突变,但不同的是,其DNA序列中碱基对变化不同,为ATG→ATA[20],或位于不同的外显子上[21]。本研究中位于PSEN1基因第5外显子的139号密码子的氨基酸突变,为国内首次发现的PSEN1基因突变位点。
PSEN1基因位于14q24.2号染色体上,其编码蛋白是γ-分泌酶的一部分,当PSEN1基因发生突变,会引起γ-分泌酶对APP切割异常,导致Aβ42/Aβ40比值的增加[22]。Aβ42比Aβ40更容易在脑内形成淀粉样聚集,最终导致AD的发生[23]。PSEN1在促进和维持记忆以及神经元存活方面可能起着重要作用[24-25]。与APP突变携带者及PSEN2突变携带者相比,PSEN1突变的病人通常发病年龄早,症状开始年龄平均比APP突变携带者早8.4岁(分别为42.9岁和51.3岁),比PSEN2突变携带者(57.1岁)早14.2岁[19]。有研究显示,EOAD始于35岁之前,几乎完全由PSEN1突变引起[26-27]。本家系中病人Ⅲ2136岁发病,Ⅲ1342岁发病,Ⅲ1743岁发病,均属于EOAD,符合FAD发病年龄小的特点。除了典型的认知功能下降外,PSEN1基因突变相关的AD病人在疾病发展过程中还可出现癫痫和肌阵挛、痉挛性截瘫、帕金森综合征和小脑共济失调等锥体外系表现,这是常染色体显性遗传EOAD的共同特征[28],而本文报道的家系成员仅出现了认知功能减退及精神症状,尚未发现癫痫发作及锥体外系症状,仍需进一步随访观察。
本研究新发现的突变p.M139I在国内为首次报道,扩大了PSEN1基因突变谱。中国人口虽然众多,但关于FAD家系的报道及相应突变位点的研究甚少。本文报道的AD家系病人疾病发展迅速,病程短,很快出现死亡,对于拓展遗传性FAD研究思路具有重要的意义,后续我们将针对此新突变位点进行基因功能分析。
[参考文献]
[1] QUERFURTH H W, LAFERLA F M. Alzheimers disease[J]. New England Journal of Medicine, 2010,362(4):329.
[2] BERTRAM L, LILL C M, TANZI R E. The genetics of Alzheimer disease: back to the future[J]. Neuron, 2010,68(2):270-281.
[3] SHERRINGTON R, ROGAEV E I, LIANG Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimers disease[J]. Nature, 1995,375(6534):754-760.
[4] LEVY-LAHAD E, WASCO W, POORKAJ P, et al. Candidate gene for the chromosome 1 familial Alzheimers disease locus[J]. Sci N Y N Y, 1995,269(5226):973-977.
[5] GOATE A, CHARTIER-HARLIN M C, MULLAN M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial[J]. Nature, 1991,349(6311):704-706.
[6] RADEMAKERS R, CRUTS M, VAN BROECKHOVEN C. Genetics of early-onset Alzheimer dementia[J]. The Scientific World Journal, 2003,3(3):497-519.
[7] 馬爱军,郭晓军,韩莹,等. 早发性Alzheimer病一家系六例[J]. 中华医学遗传学杂志, 2014,31(5):674-675.
[8] CASTELLANI R J, ROLSTON R K, SMITH M A. Alzheimer disease[J]. Disease A Month, 2010,56(9):484-546.
[9] GALLAWAY P J, MIYAKE H, BUCHOWSKI M S, et al. Physical activity: a viable way to reduce the risks of mild cognitive impairment, Alzheimers disease, and vascular dementia in older adults[J]. Brain Sci, 2017,7(2):22.
[10] MARTNEZ G, VERNOOIJ R W, FUENTES PADILLA P, et al. 18F PET with florbetaben for the early diagnosis of Alzheimers disease dementia and other dementias in people with mild cognitive impairment (MCI)[J]. Cochrane Database Syst Rev, 2017,11:CD012883.
[11] HERHOLZ K, SALMON E, PERANI D, et al. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET[J]. NeuroImage, 2002,17(1):302-316.
[12] CHTELAT G, EUSTACHE F, VIADER F, et al. FDG-PET measurement is more accurate than neuropsychological assessments to predict global cognitive deterioration in patients with mild cognitive impairment[J]. Neurocase, 2005,11(1):14-25.
[13] REITZ C, MAYEUX R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers[J]. Biochemical Pharmacology, 2014,88(4):640-651.
[14] CHOURAKI V, SESHADRI S. Genetics of Alzheimers di-sease[J]. Biomed Research International, 2013,2013(6326):1410.
[15] LPEZ-RIQUELME N, ALOM-POVEDA J, VICIANO-MOROTE N, et al. Apolipoprotein E ε4 allele and malondialdehyde level are independent risk factors for Alzheimers di-sease[J]. SAGE Open Medicine, 2016,4:205031211562673.
[16] GENIN E, HANNEQUIN D, WALLON D, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheri-tance[J]. Mol Psychiatry, 2011,16(9):903-907.
[17] POTTIER C, RAVENSCROFT A, BROWN P H, et al. TYROBP genetic variants in early-onset Alzheimers disease[J]. Neurobiology of aging, 2016,48(2):9-15.
[18] GUERREIRO R J, LOHMANN E, KINSELLA E, et al. Exome sequencing reveals an unexpected genetic cause of di-sease: NOTCH3 mutation in a Turkish family with Alzhei-mers disease[J]. Neurobiology of Aging, 2012,33(5):1008.e17-1008.e23.
[19] CRUTS M, THEUNS J, VAN BROECKHOVEN C. Locus-specific mutation databases for neurodegenerative brain diseases[J]. Hum Mutat, 2012,33(9):1340-1344.
[20] BOTEVA K, VITEK M, MITSUDA H, et al. Mutation ana-lysis of presenillin 1 gene in Alzheimers disease[J]. Lancet Lond Engl, 1996,347(8994):130-131.
[21] KIM H J, KIM H Y, KI C S, et al. Presenilin 1 gene mutation (M139I) in a patient with an early-onset Alzheimers disease: clinical characteristics and genetic identification[J]. Neurol Sci: Off J Italian Neurol Soc Italian Soc Clin Neurophysiol, 2010,31(6):781-783.
[22] SUN L F, ZHOU R, YANG G H, et al. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase[J]. Proceedings of the National Academy of Sciences of the United States of America, 2017,114(4):E476-E485.
[23] JAN A, GOKCE O, LUTHI-CARTER R, et al. The ratio of monomeric to aggregated forms of Aβ40 and Aβ42 is an important determinant of amyloid-β aggregation, fibrillogenesis, and toxicity[J]. Journal of Biological Chemistry, 2008,283(42):28176-28189.
[24] LEE S, SHARMA M, SUDHOF T C, et al. Synaptic function of nicastrin in hippocampal neurons[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014,111(24):8973-8978.
[25] TABUCHI K, CHEN G Q, SUDHOF T C, et al. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration[J]. The Journal of Neuroscience, 2009,29(22):7290-7301.
[26] HOLMES C. Genotype and phenotype in Alzheimers disease[J]. Br J Psychiatry: J Ment Sci, 2002,180(2):131-134.
[27] CAMPION D, BRICE A, DUMANCHIN C, et al. A novel presenilin 1 mutation resulting in familial Alzheimers disease with an onset age of 29 years[J]. Neuroreport, 1996,7(10):1582-1584.
[28] RUDZINSKI L A, FLETCHER R M, DICKSON D W, et al. Early onset familial Alzheimer disease with spastic paraparesis, dysarthria, and seizures and N135S mutation in PSEN1[J]. Alzheimer Dis Assoc Disord, 2008,22(3):299-307.
(本文編辑 马伟平)
