玉米秸秆低温降解复合菌系降解能力及微生物组成研究*
2020-11-10青格尔于晓芳高聚林王志刚胡万吉闹干朝鲁胡树平孙继颖屈佳伟
青格尔, 于晓芳**, 高聚林**, 王志刚, 胡万吉, 闹干朝鲁, 王 振, 胡树平, 孙继颖, 屈佳伟
玉米秸秆低温降解复合菌系降解能力及微生物组成研究*
青格尔1,2, 于晓芳1,2**, 高聚林1,2**, 王志刚1,2, 胡万吉1, 闹干朝鲁2,3, 王 振2,3, 胡树平2,4, 孙继颖1,2, 屈佳伟1,2
(1. 内蒙古农业大学农学院 呼和浩特 010019; 2. 内蒙古自治区作物栽培与遗传改良重点实验室 呼和浩特 010019; 3. 内蒙古农业大学园艺与植保学院 呼和浩特 010019; 4. 内蒙古农业大学职业技术学院 包头 014100)
根据还田秸秆配施尿素的生产实际, 对玉米秸秆低温高效降解复合菌系GF-20进行氮源培养基驯化, 探明其菌种组成和功能多样性及其与菌源菌种结构差异, 完善复合菌系筛选方法, 促进其开发利用。本文以低温高效降解复合菌系GF-20为研究对象, 在硫酸铵和尿素不同配比下连续继代培养10代, 获得不同氮源菌系(硫酸铵处理N1, 硫酸铵和尿素混合处理N2-N5, 尿素处理N6), 测定其玉米秸秆降解率, 评价复合菌系秸秆降解效率; 采用MiSeq高通量测序对菌源土壤样品及不同氮源下继代培养的复合菌系菌种组成和功能多样性进行研究。结果显示N2处理玉米秸秆降解率显著高于其他处理; 菌源土壤的Alpha多样性指数显著高于继代培养后的复合菌系, 不同处理间N2处理显著高于其他氮处理; 菌源和复合菌间以及不同氮处理间菌种组成具有显著差异, N2处理菌种组成多样性较高, 菌群结构更加丰富、均匀, 且碳水化合物的代谢通路丰度较高。菌源经限制性继代筛选后得到了参与玉米秸秆降解过程的功能菌, 能有效提高秸秆降解率, 其在硫酸铵和尿素氮源为0.16%+0.04%的条件下, 菌系的秸秆降解效率较高, 这为复合菌的生产实际开发利用提供了理论依据。
菌源; 复合菌系; 氮源; 高通量测序; 菌种组成多样性; 玉米秸秆降解
秸秆还田是提高秸秆资源综合利用率、平衡农田生态系统和实现农业可持续发展的有效措施[1]。由于玉米()秸秆木质纤维素的结晶区域组成, 使得还田玉米秸秆难以降解[2]; 而北方高寒地区秋冬季低温、干燥等气候因素进一步限制了还田玉米秸秆的有效腐解。因此, 在低温条件下施加外源菌剂加速分解木质纤维素对玉米秸秆资源的开发利用具有重要意义。根据秸秆酶解过程以及微生物间的协同关系, 直接从自然环境中筛选或组配秸秆高效降解复合菌群, 用以提高还田秸秆的降解效率是目前秸秆高效降解的有效途径之一[3-4]。其中, 菌源的选择、复合菌群的筛选驯化方法对于获得秸秆高效降解微生物成败至关重要。前人研究表明, 根据应用目的从相似的生态环境中可筛选到更高效适宜的菌种[5]。Li等[6]和Feng等[7]以高温期堆肥、腐烂作物秸秆和秸秆还田土壤为菌源, 筛选得到一系列中高温复合菌系, 其秸秆降解率达30%~50%, 被应用于堆肥腐熟或南方高温区秸秆还田。
本研究团队为了获得玉米秸秆低温高效降解菌以用于北方低温种植区玉米秸秆原位促腐还田, 从低温(−7~8 ℃)生态环境中采集多年秸秆还田土壤作为玉米秸秆低温高效降解复合菌系筛选的菌源材料,经长期的碳源限制性继代培养与低温驯化培养获得一组微生物复合菌系GF-20[8], 其实验室秸秆降解率达30%。但菌源材料与复合菌系GF-20间的菌种组成相关性及不同尿素含量处理对菌种组成的差异性尚不明确。这无疑关系到该菌系秸秆高效降解机理的揭示, 以及其能否被继续开发、应用于生产实际。因此, 本研究以菌源样品、筛选得到的复合菌系GF-20及其尿素氮源驯化后的系列菌系为材料, 通过对各复合菌系菌种多样性、玉米秸秆分解能力和菌群结构特性等指标的测定及分析, 揭示复合菌系GF-20与菌源材料间, 以及不同尿素氮源含量驯化得到的复合菌系间的菌种异同, 通过与COG(蛋白质直系同源簇)和KEGG(代谢通路)数据库比对及菌株功能性分析从而确定复合菌系中的关键功能菌,以及菌系适宜的尿素氮源条件, 为该复合菌系的开发利用提供理论依据和技术支持, 也给其他高效降解菌系的筛选和利用提供借鉴。
1 材料与方法
1.1 试验时间与地点
试验于2018—2019年在内蒙古农业大学玉米中心微生物实验室(内蒙古包头市土默特右旗沟门镇)进行。
1.2 供试材料
菌源土壤(编号为Q, 为本文复合菌系GF-20的筛选菌源)取自内蒙古通辽市常年秸秆还田土壤(43.37°N、122.16°E, 海拔400 m, 年平均气温5.5 ℃)和吉林省长春市连续秸秆还田土壤(43.82°N、125.32°E, 海拔250 m, 年平均气温4.8 ℃)的混合样品, 供试复合菌系为本实验室筛选驯化的玉米秸秆降解复合菌系GF-20(编号为N1)[8]。玉米秸秆取自内蒙古农业大学蒙西综合试验站(内蒙古包头市土默特右旗沟门镇)试验田收获的玉米秸秆(C/N为48.98, 纤维素、半纤维素和木质素含量分别为51.24%、32.72%和9.15%, 全氮6.67 g·kg-1, 全磷2.78 g·kg-1, 全钾11.48 g·kg-1), 洗净烘干后剪成1~2 cm短节。滤纸选用Whatman No.Ⅰ滤纸, 裁成1 cm×10 cm大小。
1.3 培养条件
以基础培养基[(NH4)2SO42.0 g、K2HPO41.0 g、MgSO4·7H2O 0.05 g、CaCO32.0 g、NaCl 0.2 g、蒸馏水1 L]为继代培养基, 复合菌以5%~10%(/)的接种量接入装有40 mL基础培养基和1 g玉米秸秆碳源的100 mL三角瓶中, 置于10 ℃恒温条件下静置培养[9]。
1.4 试验设计
基础培养基中氮源设硫酸铵+尿素, 含量分别为0.16%+0.04%(N2)、0.12%+0.08%(N3)、0.08%+0.12%(N4)、0.04%+0.16%(N5)、0+0.2%(N6)。对复合菌系GF-20进行尿素驯化处理, 原始培养基为硫酸铵0.2%+尿素0 (N1), 在以上处理条件下连续继代培养10代以上, 获得不同菌系, 每处理均3次重复。
1.5 主要测定指标与方法
1)复合菌系玉米秸秆降解率的测定。将所得复合菌系以5%的接种量接入玉米秸秆培养基[9]中, 以不接菌的为对照(CK), 在10 ℃恒温条件下培养15 d和30 d后, 采用失重法测定玉米秸秆降解率, 3次重复。秸秆降解率(%)=(0-1)/0×100%。0表示接种前培养基中的秸秆重(g),1表示培养结束烘干后降解剩余物重(g)。
2)MiSeq 16S rDNA扩增子测序。采用土壤基因组DNA提取试剂盒和细菌基因组DNA提取试剂盒(中国, 天根生化科技有限公司)提取菌源土壤和各复合菌系的基因组DNA, 利用1%琼脂糖凝胶电泳检测。获得的基因组DNA由北京奥维森基因科技有限公司(Allwegene Tech. Co. Ltd., Beijing)完成MiSeq高通量测序, 对获得的数据通过序列拼接、过滤和去嵌合体得到优化序列, 进行OTU(Operational Taxonomic Units)聚类及各分类水平注释。
3)功能注释。基于测序得到的序列, 与COG(蛋白质直系同源簇)和KEGG(代谢通路)数据库进行比对[10]。
4)数据统计分析。利用QIIME(v1.8.0)软件以97%相似度水平将序列进行聚类物种分类的OTU, 获得物种分类水平的比例信息及不同分类水平的群落结构; 通过Uclust(Version 1.8)、Usearch(Version 8.1.1861)等数据库进行物种比对, 得到各水平的分类信息, 进行样本组成及样本间群落结构差异分析; 采用R(v3.4.4)软件绘制各样品在各分类水平的比较图, 基于聚类结果, 进行Alpha多样性分析和Beta多样性分析。
2 结果与分析
2.1 不同氮源下复合菌系玉米秸秆降解率
不同氮源条件下复合菌系GF-20的玉米秸秆降解率如图1所示。不同氮源处理降解率均显著高于CK; 不同处理随着尿素比例的增加, 复合菌系GF-20玉米秸秆降解率呈先上升后下降趋势。培养至15 d和30 d, 均以N2条件下复合菌系的玉米秸秆降解率最高, 分别为22.64%和37.58%, 较CK高15.85%和21.43%, 且显著高于其他处理。

图1 不同氮源处理复合菌系玉米秸秆降解率
N1、N2、N3、N4、N5和N6的氮源为硫酸铵+尿素, 比例为分别0.2%+0、0.16%+0.04%、0.12%+0.08%、0.08%+0.12%、0.04%+0.16%、0+0.2%。CK为不加菌对照, 培养条件同N1。不同小写字母表示不同处理间差异显著。The nitrogen resources of N1, N2, N3, N4, N5 and N6 are ammonium sulfate+urea with ratios of 0.2%+0, 0.16%+0.04%, 0.12%+0.08%, 0.08%+0.12%, 0.04%+0.16%, and 0+0.2%, respectively. CK is non-inoculated microbial consortium (control), and the culture conditions are the same as N1. Different lowercase letters mean significant differences among different treatments.
2.2 菌源样品及不同氮源下复合菌系Alpha多样性指数分析
所有样品及处理覆盖深度指数均在0.99以上, 说明本次测序结果基本能代表样品的真实情况。菌源土壤平均OUT数为1 705, 显著高于筛选获得的复合菌系, 处理N1-N6平均OUT数为58; 由Shannon、PD whole tree、Observed species和Chao指数可知, 菌源土壤显著高于处理N1-N6; 不同氮源培养基获得的复合菌系表现为混合氮源处理(N2-N5)高于单一氮源处理(N1, N6), 且N2处理显著高于其他处理, 其Shannon、PD whole tree、Observed species和Chao多样性指数分别为4.36、6.14、59.93和62.45(图2)。
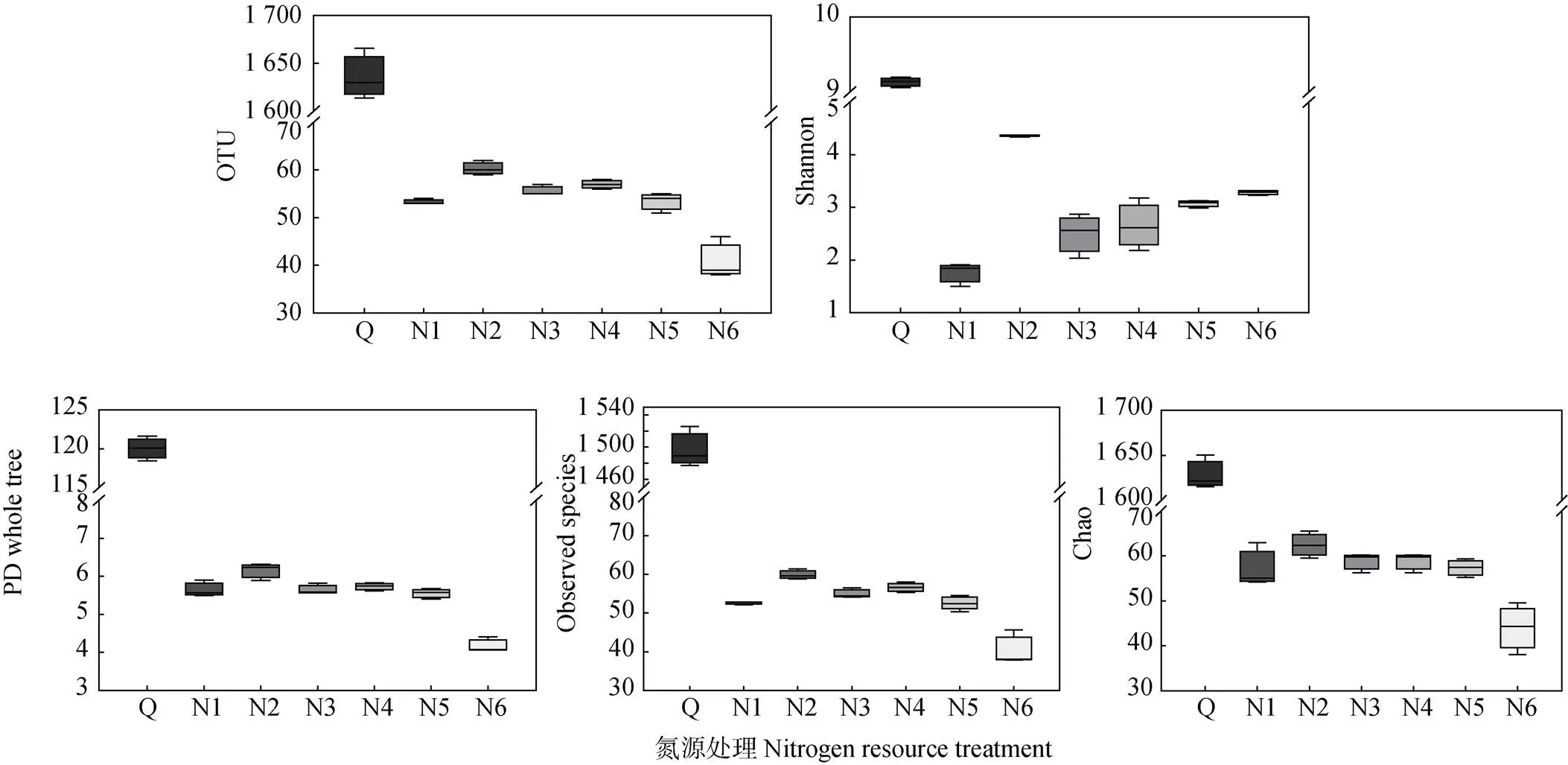
图2 菌源样品及不同氮源处理复合菌系Alpha多样性指数
Q为菌源土壤。N1、N2、N3、N4、N5和N6的氮源为硫酸铵+尿素, 比例为分别0.2%+0、0.16%+0.04%、0.12%+0.08%、0.08%+0.12%、0.04%+0.16%、0+0.2%。Q is original microbe source. The nitrogen resources of N1, N2, N3, N4, N5 and N6 are ammonium sulfate+urea with ratios of 0.2%+0, 0.16%+0.04%, 0.12%+0.08%, 0.08%+0.12%, 0.04%+0.16%, and 0+0.2%, respectively.
2.3 菌源样品及不同氮源下复合菌系细菌群落结构分析
对所有样品测序结果进行物种比对, 得到各水平的分类信息。门分类水平, 变形菌门(Proteobacteria)在所有样本中丰富度最高(图3a)。菌源样品中变形菌门丰度为36.98%, 其他优势门是放线菌门(Actinobacteria)和绿弯菌门(Chloroflexi), 丰度分别为24.06%和10.64%; 复合菌系GF-20中变形菌门丰度为85.52%, 拟杆菌门(Bacteroidetes)丰度为13.86%。纲分类水平, 在所有样本中α变形菌(Alphaproteobacteria)β变形菌(Betaproteobacteria)和γ变形菌(Gammaproteobacteria)丰度较高。而黄杆菌纲(Flavobacteriia)在复合菌中丰富度较高, 在菌源中仅有0.32%(图3b)。属分类水平(图4), 菌源主要由(5.00%)、(3.73%)、(3.13%)、节细菌属(, 2.26%)、(2.24%)、壤霉菌属(, 2.01%)组成, 复合菌系主要由纤维弧菌属()固氮螺菌属()黄杆菌属()土地杆菌属()假单孢菌属()纤维菌属()等组成, 且不同氮源处理丰度具有显著差异。说明复合菌系及菌源样品的菌群结构差异较大, 长期限制性继代培养及低温驯化培养显著影响菌系菌种组成, 大量菌属被淘汰。
不同氮源处理间, 在门分类水平, N1、N2和N6中拟杆菌门丰度较高, 为13.86%、37.01%和17.72%; 放线菌门在N2-N5中含量分别为1.18%、5.71%、6.29%和26.11%, 表现为随着尿素含量的增加而增加。属水平, N1-N6处理复合菌系的细菌菌群结构差异显著(图3c, 图4), 复合菌系GF-20在N1条件下的优势菌属为纤维弧菌属(74.54%), 其次为固氮螺菌属(7.40%); N2的优势菌属为纤维弧菌属(22.24%)、黄杆菌属(9.35%)、土地杆菌属(8.60%)和norank_c__(9.03%); N3主要由纤维弧菌属(14.3%)、假单胞菌属(55.72%)和纤维菌属(5.48%)组成; N4主要由假单胞菌属(56.82%)、纤维菌属(5.80%)、德沃斯氏菌属(, 5.44%)、芽殖杆菌属(, 4.57%)和纤维弧菌属(4.44%)组成; N5主要由纤维菌属(25.98%)、(28.11%)、食酸菌属(, 10.97%)和噬氢菌属(, 6.36%)组成; N6的优势菌属为纤维弧菌属(31.13%)、短波单胞菌属(, 18.5%)、黄杆菌属(12.8%)和无色杆菌属(12.56%)等。可见尿素含量对复合菌系GF-20的细菌菌群结构影响较大, 培养基中加入尿素后使优势菌种种类增加, 混合氮源效果尤其明显。综上所述可知, 菌源样品和所筛选获得的复合菌系, 以及不同氮源下的菌系间菌种组成差异显著, 主要体现在放线菌门、拟杆菌门、变形菌门的纤维弧菌属、假单胞菌属、、黄杆菌属、无色杆菌属、纤维菌属等参与秸秆降解微生物丰度的差异。

图3 菌源样品及不同氮源处理复合菌系门(a)和纲(b)水平群落结果及属水平(c)Heatmap图分析
将在所有样本中丰度占比均小于0.02%的物种归为Others。Q为菌源土壤。N1、N2、N3、N4、N5和N6的氮源为硫酸铵+尿素, 比例分别为0.2%+0、0.16%+0.04%、0.12%+0.08%、0.08%+0.12%、0.04%+0.16%和0+0.2%。Microbes with less than 0.02% abundance in all samples are classified as others. Q is original microbe source. The nitrogen resources of N1, N2, N3, N4, N5, and N6 are ammonium sulfate+urea with ratios of 0.2%+0, 0.16%+0.04%, 0.12%+0.08%, 0.08%+0.12%, 0.04%+0.16% and 0+0.2%, respectively.
2.4 不同氮源下复合菌系菌群群落结构Heatmap分析
根据所有样品在属水平的物种注释及丰度信息, 选取丰度top 20的属, 绘制属水平的Heatmap图(图3c)。在属水平上, 不同氮处理复合菌系优势菌属不同, 混合氮源处理N2-N5的优势属数量多于单一氮源N1和N6处理, N1处理中的固氮螺菌属等属丰度在N6中显著降低, 而丰度较低的如无色杆菌属短波单胞菌属噬氢菌属根瘤菌属()贪噬菌()等属在混合氮源处理中成为优势属。
由图3c和图4可知, 复合菌系GF-20在N1条件下, 纤维弧菌属的丰度为74.50%, 但在含有不同尿素比例氮源条件下继代培养数量显著变化, 尤其在混合氮源条件下随着尿素含量的增加而逐渐减少至0.81%; 德沃斯氏菌属在N1条件下丰度为0.99%, 但在N4处理中增加至5.61%; 黄杆菌属、地杆菌属、短波单胞菌属、无色杆菌属等在N1条件下丰度均很低, 但在含有尿素氮源的菌系中大量存在, 成为优势菌属。说明限制性继代培养是有选择性的进行筛选, 大量富集与秸秆纤维素降解相关的菌属, 提高菌属数量比, 从而促进秸秆的降解。纤维菌属在N1条件下丰度仅仅为0.03%, 但在混合氮源处理下含量逐渐增加, 在N5处理下含量达27.31%; 假单胞菌属在N1条件下含量为0.04%, 但在混合氮源N3和N4处理下数量剧增至57.68%和59.25%; 无色杆菌属和鞘脂单胞菌属()在N1处理中为0.08%和0.06%, 但随着尿素含量的增加, 含量逐渐增加, N6处理增加至12.84%和3.59%;和在N1处理中含量为0.70%和0.14%, 在混合氮源N2处理中增加至3.91%和2.35%;在混合氮源处理N5中含量最高, 为29.75%;在N1处理中含量为1.68%, 在处理N2中增加至5.42%, 随着尿素氮源的增加含量逐渐降低, 在N6处理中减少至0; 固氮螺菌属和金黄杆菌属()在N1处理中的丰度分别为7.2%、3.78%和2.86%, 但随着尿素氮源的增加, 其含量逐渐减少, 尿素处理N6的含量减少至0。由此可知, 硫酸铵和尿素含量对复合菌系GF-20繁殖有明显的影响, 由Alpha多样性指数及菌群组成(图2, 图4)可知, N2处理中富集了更多的参与秸秆降解的微生物, 菌种组成多样性较高, 菌群结构更加丰富。
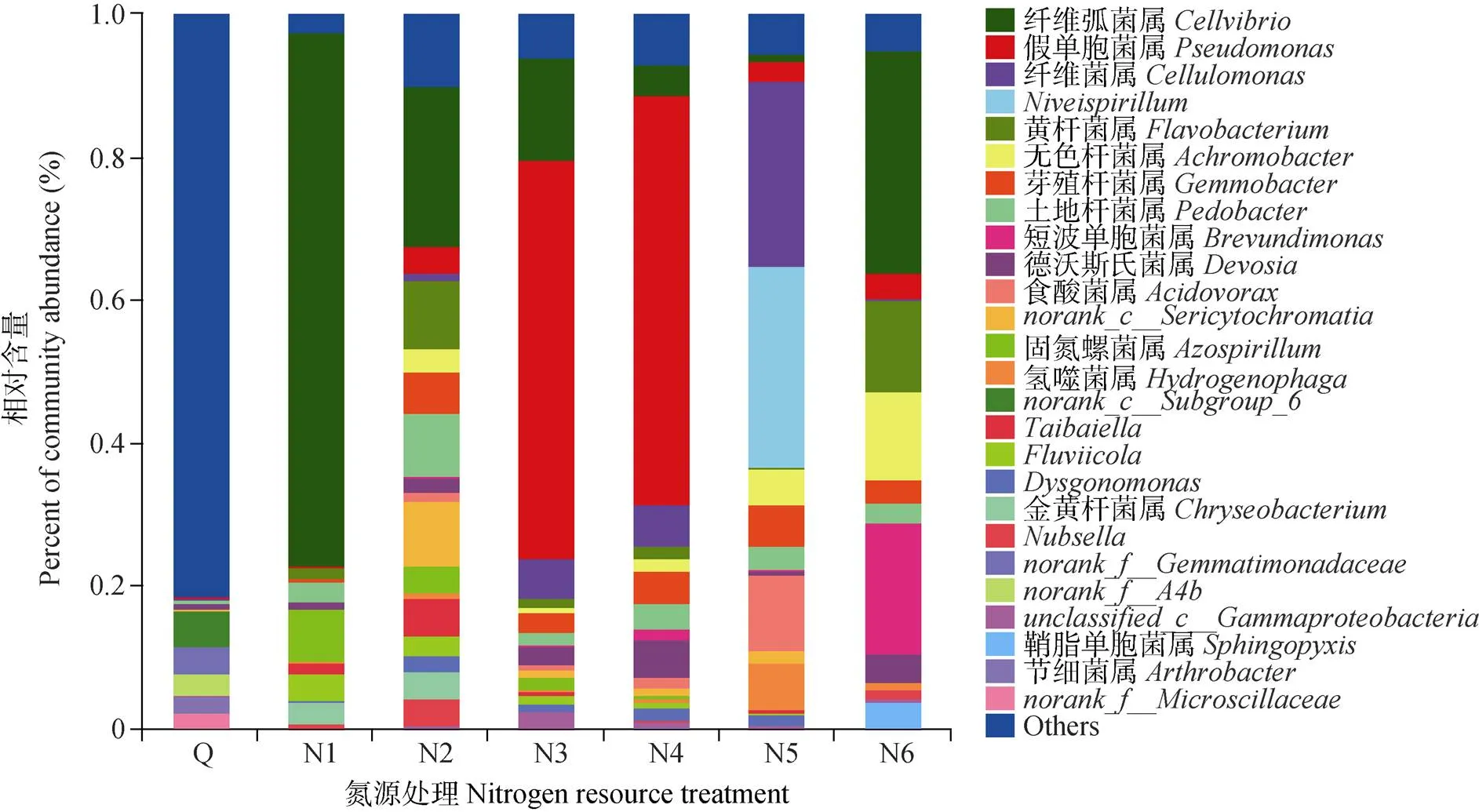
图4 菌源及不同氮源处理复合菌系细菌群落结构分布(属水平)
Q为菌源土壤。N1、N2、N3、N4、N5和N6的氮源为硫酸铵+尿素, 比例分别为0.2%+0、0.16%+0.04%、0.12%+0.08%、0.08%+0.12%、0.04%+0.16%、0+0.2%。Q is original microbe source. The nitrogen resources of N1, N2, N3, N4, N5 and N6 are ammonium sulfate+urea with ratios of 0.2%+0, 0.16%+0.04%, 0.12%+0.08%, 0.08%+0.12%, 0.04%+0.16%, 0+0.2%, respectively.
2.5 不同氮源下复合菌系菌群结构PCoA分析及Rank Abundance分析
根据不同氮源处理菌种组成及进化关系进行PCoA分析, 结果表明样品分布较为分散(图5a), 说明不同氮源处理菌系间物种丰度及差异度均存在差异, 其中GF-20(N1)、N2及N6与N4和N5处理的距离较远, 而N3、N4和N5的距离较近, 再次说明不同氮源处理, 显著影响复合菌系菌种物种组成, 且相同氮源的组成菌群结构更加相似。Rank-Abundance说明物种丰富度和均匀度。由图5b可知, N2处理水平方向曲线宽度最宽且平滑, 结合OTU指数分析发现, N2处理OUT数为60±0.88, 显著高于其他处理, 较N1及N3-N6分别高13.12%、8.38%、5.84%、13.12%和47.15%(图2), 说明N2处理物种组成更加丰富和均匀。
2.6 不同氮源下复合菌系菌群Venn图分析
进一步在属水平进行Venn图分析, 统计样本中所共有和独有菌属数目, 直观体现各样本的菌属组成相似性及重叠情况。由图6可知, 有32个属为6个处理共有的, 被称为核心微生物, 直接或间接参与玉米秸秆降解代谢, 主要由放线菌、α变形菌、变形菌、拟杆菌纲的纤维弧菌属、假单胞菌属、黄杆菌属、纤维菌属、德沃斯氏菌属、无色杆菌属等组成。
2.7 不同氮源下复合菌系功能多样性预测分析
将不同氮源处理复合菌系测序获得的序列与COG和KEGG数据库进行系统功能预测分析。通过COG分析对潜在功能进行分类, 结果表明amino acid transport and metabolism (氨基酸运输/代谢, E)、general function prediction only (一般功能预测, R)、signal transduction mechanisms (信号转导机制, T)、function unknown (未知功能, S)的注释丰度最多为主导功能(图7)。将24个类别概括为4类: 信息存储与处理(第1类)、细胞过程和信号传导(第2类)、代谢(第3类)以及功能较差类别(第4类), 其中carbohydrate transport and metabolism(碳水化合物转运和代谢)的相对丰度在N1和N2处理明显高于其他处理, 说明其通过增加碳水化合物的运输和代谢功能, 促进秸秆的降解。

图5 不同氮源处理复合菌系属水平PCoA (a)和RankAbundance (b)分析
N1、N2、N3、N4、N5和N6的氮源为硫酸铵+尿素, 比例分别为0.2%+0、0.16%+0.04%、0.12%+0.08%、0.08%+0.12%、0.04%+0.16%和0+0.2%。The nitrogen resources of N1, N2, N3, N4, N5 and N6 are ammonium sulfate+urea with ratios of 0.2%+0, 0.16%+0.04%, 0.12%+0.08%, 0.08%+0.12%, 0.04%+0.16%, and 0+0.2%, respectively.

图6 不同氮源处理复合菌系属水平菌种分布比较的Venn图分析
N1、N2、N3、N4、N5和N6的氮源为硫酸铵+尿素, 比例分别为0.2%+0、0.16%+0.04%、0.12%+0.08%、0.08%+0.12%、0.04%+0.16%和0+0.2%。The nitrogen resources of N1, N2, N3, N4, N5 and N6 are ammonium sulfate+urea with ratios of 0.2%+0, 0.16%+0.04%, 0.12%+0.08%, 0.08%+0.12%, 0.04%+0.16%, and 0+0.2%, respectively.
通过KEGG数据库比对(表1)发现, metabolism (代谢)功能注释到的结果最多, 占全功能分类的60%以上, 不同处理间N2处理明显高于其他处理。为进一步分析代谢途径, 在KEGG第3分类水平进行分析, 选取丰度大于1.00%的路径进行统计(图8), 共获得21条代谢路径, ABC transporters(ABCT)和two-component system(TCSP)注释最多,其中9条代谢途径丰度呈N2大于其他处理, 其中包含fructose and mannose metabolism(果糖和甘露糖代谢)、starch and sucrose metabolism(淀粉和蔗糖代谢)和amino sugar and nucleotide sugar metabolism(氨基糖和核苷酸糖代谢)等相关碳水化合物的代谢路径。再次说明复合菌系在氮源为硫酸铵和尿素含量为0.16+0.04% (N2)条件下, 增加碳水化合物代谢功能, 促进秸秆物质的降解。

图7 不同氮源处理复合菌系COG功能注释相对丰度
N1、N2、N3、N4、N5和N6的氮源为硫酸铵+尿素, 比例分别为0.2%+0、0.16%+0.04%、0.12%+0.08%、0.08%+0.12%、0.04%+0.16%和0+0.2%。The nitrogen resources of N1, N2, N3, N4, N5 and N6 are ammonium sulfate+urea with ratios of 0.2%+0, 0.16%+0.04%, 0.12%+0.08%, 0.08%+0.12%, 0.04%+0.16%, and 0+0.2%, respectively.

表1 不同氮源处理复合菌系KEGG注释结果的第2类水平统计

续表1
N1、N2、N3、N4、N5和N6的氮源为硫酸铵+尿素, 比例分别为0.2%+0、0.16%+0.04%、0.12%+0.08%、0.08%+0.12%、0.04%+0.16%和0+0.2%。RA%指相对丰度, log指注释数量的log10。The nitrogen resources of N1, N2, N3, N4, N5 and N6 are ammonium sulfate+urea with ratios of 0.2%+0, 0.16%+0.04%, 0.12%+0.08%, 0.08%+0.12%, 0.04%+0.16%, 0+0.2%, and respectively. RA% refers to relative abundance, and log refers to log10 of the number of annotations.
3 讨论
本研究中菌源材料取自内蒙古通辽市常年秸秆还田和吉林长春市连续秸秆还田混合土壤样品, 主要由放线菌(24.06%)、变形菌(36.98%)、酸杆菌(9.71%)、绿弯菌(10.64%)、拟杆菌(6.94%)、芽单胞菌(5.74%)等门组成, 富含降解纤维素、半纤维素、木质素的菌属, 如鞘氨醇单胞菌属(, 0.99%)、芽孢杆菌属(, 0.22%)、梭菌属(, 0.1%)、纤维弧菌属(, 0.01%)、(0.02%)、纤维菌属(, 0.07%)、(0.04%)、(0.14%)等, 从菌源样品中经长期限制性继代培养筛选出了玉米秸秆低温高效降解菌系GF-20, 其在低温10 ℃下的秸秆降解率达30%。复合菌系筛选驯化的最终目的是应用到秸秆还田土壤中, 通常以生态条件与目标菌系相近的常年秸秆还田土壤作为菌源筛选材料, 提高筛选效率及应用效果。

图8 不同氮源处理复合菌系KEGG主要代谢途径的相对丰度(>1.00%)
N1、N2、N3、N4、N5和N6的氮源为硫酸铵+尿素, 比例分别为0.2%+0、0.16%+0.04%、0.12%+0.08%、0.08%+0.12%、0.04%+0.16%和0+0.2%。The nitrogen resources of N1, N2, N3, N4, N5 and N6 are ammonium sulfate+urea with ratios of 0.2%+0, 0.16%+0.04%, 0.12%+0.08%, 0.08%+0.12%, 0.04%+0.16%, and 0+0.2%, respectively.
3.1 限制性继代培养过程中保留菌属分析
限制性继代培养筛选过程中保留了在低温条件下直接或间接参与木质纤维素降解代谢途径的菌, 菌源样品及复合菌系数量比高于0.1%的共有菌属有9个(图3, 图4, 图6), 其中纤维弧菌属可分泌3~4种纤维素降解酶系, 具有木聚糖酶基因, 降解羧甲基纤维素、纤维素、半纤维素、几丁质等, 如混合纤维弧菌(subsp.)、J3-8、等[11-12], 其中混合纤维弧菌属于产纤维素酶类的兼性厌氧微生物, 分泌的纤维素酶属于典型的低温纤维素酶[13]; 土地杆菌属和黄杆菌属大多源自于土壤, 可利用秸秆降解中间代谢产物促进秸秆分解, 如sp.可分泌葡糖苷酶、糖苷水解酶, 可利用碳水化合物、醇类和糖苷类等[14-15],sp.sp.分泌过氧化氢酶、氧化酶、葡糖苷酶、半乳糖苷酶等, 利用丙酸、乳酸等秸秆降解中间产物, 消除底物反馈抑制作用[16-17]; 复合菌系中还含有鞘氨醇单胞菌属根瘤菌氢噬菌属柄杆菌属()等含量低但起到关键作用的菌, 其中鞘氨醇单胞菌属和鞘脂菌属()是降解多环芳烃(PAHs)的重要功能微生物, 如sp. GD542参与石油降解途径[18],分泌过氧化氢酶和氧化酶, 代谢产生丙酸、琥珀酸、L-丙氨酰胺等有机酸类, 在秸秆分解初期调节培养体系pH值[19]; 氢噬菌属通常分泌水解酶, 如为兼氧细菌, 分泌氧化酶[20]; La等[21]报道sp.等菌分泌氧化酶、过氧化氢酶, 促进秸秆物质的分解; 德沃斯氏菌属的sp.等分泌过氧化氢酶, 利用木糖、戊醛糖、纤维素等[22-23]; 柄杆菌属中的能降解水杨苷[24],分泌糖苷酶、均可在4 ℃低温条件下生长, 分泌水解酶类,以纤维二糖、木糖为碳源产酸, 分泌葡萄糖苷酶[25]; 根瘤菌属大多存在于根瘤菌中, 既可合成纤维素也降解木质素, 其中YS-1r具有木质素降解能力, 能够降解各种木质素单体、二聚体以及柳枝草和紫花苜蓿的天然木质素, 并且分泌木质素过氧化物酶(LiP)[26-27]。由此可知, 复合菌系GF-20中保留了菌源土壤中的功能菌, 通过限制性培养使其大量扩繁, 大大提高丰度, 从而促进玉米秸秆的降解。
3.2 复合菌系菌种功能多样性分析
复合菌系的限制性继代筛选过程既是淘汰与秸秆降解无关或不适应培养基条件的菌属过程, 也是富集木质纤维素降解菌的过程。复合菌系中保留的菌属均直接或间接参与秸秆降解代谢过程, 或是分泌纤维素、半纤维素、木质素酶等直接降解木质纤维素, 如纤维弧菌属德沃斯氏菌属根瘤菌属鞘氨醇杆菌属寡养单胞菌()等; 或是分泌葡萄糖苷酶、水解酶、过氧化氢酶等消耗中间代谢产物纤维二糖、木糖、醛糖等, 促进秸秆的持续降解, 如氢噬菌属土地杆菌属黄杆菌属、柄杆菌属、、类芽孢杆菌属()等; 有些菌产生氧化酶类, 调节发酵体系酸碱度维持中性水平, 协调降解秸秆, 如鞘氨醇杆菌属黄杆菌属等分泌氧化酶类, 利用乙酸、乳酸等中间产物, 调节发酵体系酸碱度维持中性水平, 协调降解秸秆; 有些菌可能与秸秆降解没有直接关系, 是因为适应筛选培养基而得以保留。
通过COG和KEEG数据库对复合菌系功能多样性进行预测比对发现, ABCT和TSCP相对含量最多。就功能概况而言, ABCT可将氨基酸、脂质、脂糖、肽和无机离子(如金属离子)转运至细胞外部, 从细胞中摄取寡糖、多糖、单糖(例如纤维二糖、纤维糊精、葡萄糖、半乳糖和甘露糖); 而TCSP感知细胞外可溶性糖的存在, 并调节大多数糖苷水解酶(GH)和相关的ABCT, 调节糖类的转运, 以促进复杂植物材料(例如半纤维素)的利用, 并将各种结构的产物(例如木糖)转运到细胞中[28]。代谢功能注释相对丰度大多表现为N2处理含有更多的碳水化合物代谢功能, 包括果糖和甘露糖代谢、淀粉和蔗糖代谢以及氨基糖和核苷酸糖代谢等, 促进秸秆降解中间产物的代谢, 从而提高玉米秸秆木质纤维素的降解(图1); 而部分代谢基因在N6处理中注释丰度明显高于其他处理, 说明在尿素氮源条件下, 复合菌系通过促进自身代谢, 从而适应培养环境(表1)。
3.3 复合菌系间差异菌属分析
菌源土壤与其经过长时间限制性继代培养筛选驯化获得的复合菌系细菌群落结构具有显著差异, 限制性继代培养过程是微生物淘汰的过程, 具有随机性。同样, 将筛选获得的复合菌系培养于不同条件下时, 细菌群落结构也有差异, 说明菌系内部关键菌株的数量比例随着外界培养条件的变化而变化(图5, 图6)。根霉菌属()在N2中含量较高,可直接利用纤维二糖、木聚糖等参与秸秆降解代谢途径[29],而可利用纤维二糖、木糖等代谢中间产物[30]; 短波单胞菌属和无色杆菌属在N1和N2中丰度为0.05%、0.08%和0.59%、3.37%, 在N6中为18.90%和12.84%, 说明尿素为唯一氮源促进其生长,等均可分泌纤维素酶, 参与秸秆物质的降解, 短波单胞菌属的sp.sp.等可分泌葡萄糖苷酶, 参与秸秆降解代谢[31],sp.sp.等可利用纤维二糖等代谢产物; 司美茹等[32]从被石油污染的土壤和焦化污泥中分离出无色杆菌属的XL和SQ-1, 几乎能彻底降解C12−C23和C27−C43的烷烃和多环芳香烃,能氧化木糖, 具有过氧化氢酶和氧化酶活性[32-33]; 纤维菌属在混合氮源条件下丰度较高, 许多纤维菌属的细菌应用于秸秆生物降解和生物修复领域, 其中分泌的纤维素酶活性高, 包括β-葡萄糖苷酶(GH1, GH3)、内切酶(GH6, GH9)、β-1,4-葡聚糖酶(GH48, GH5)和纤维二糖磷酸化酶(GH94)[33-34];在N2中的丰度显著高于其他处理菌系,菌属中的NS114T、Y620-1等具有纤维素降解能力[34-35]。
根瘤菌属等6个丰度高于0.1%的属在N1和N2中存在, 但N6中未检测到, 可能是由于以上菌属不能以尿素为唯一氮源, 在继代过程中逐渐被淘汰或含量显著降低; 短波单胞菌属等8个属在N1处理中含量很低, 但在N6中含量显著增加, 说明尿素为唯一氮源促进以上菌种繁殖。另外,等9个属在菌源土壤中未检测到, 而在复合菌系中检测出, 说明在继代培养的条件下新增加了9个微生物属, 可能来源于空气, 随继代过程中被保留下, 或这9个微生物属在继代培养的条件下得到了大量繁殖, 数量增加, 在16S rDNA扩增测序中被检测到。菌属在N1中含量高于0.1%, 具有直接降解木质素的功能, 固氮螺菌属丰度为7.40%,与固氮螺菌属具有协同降解作用[36]; 寡养单胞菌()属于纤溶微生物, 多报道于石油降解菌种, 具有降解烷烃类物质的作用[37];可分泌脲酶和糖苷酶, 常以尿素为氮源[38]; Lewin等[39]和Abt等[40]报道的纤维素降解复合菌系中有属的; 在牛粪中富含降解纤维素和木质素的菌属[41]; 类芽孢杆菌属中的等筛选自土壤, 具有较高的纤维素酶活性, 可直接降解纤维素类物质[42];和在相关文献中暂没有报道具有秸秆或纤维素降解功能。
复合菌系通过调节微生物组成, 进一步调节其功能多样性, 从而适应不同尿素含量培养条件, 进而促进秸秆的降解。不同氮源处理间, N2处理复合菌系Alpha多样性指数显著高于其他处理, 菌种组成多样性较高,根瘤菌等参与秸秆降解途径的菌属含量增加, 促进其秸秆降解进程, 导致N2处理秸秆降解效率较高。
4 结论
菌源样品及复合菌系间的Alpha多样指数、OTU丰度和差异度、物种结构及含量均存在显著差异, 不同氮源处理间, N2处理复合菌系的菌种组成多样性较高, 菌群结构更加丰富、均匀。在限制性继代培养过程中保留含量高于0.1%菌属有纤维弧菌根瘤菌土地杆菌噬氢菌黄杆菌德沃斯氏菌柄杆菌鞘氨醇单胞菌等, 均可分泌水解酶和氧化酶, 直接或间接参与秸秆降解进程中; 在氮源硫酸铵和尿素含量0.16+0.04%(N2)配比条件下, 优势菌种为纤维弧菌、黄杆菌、土地杆菌, N2处理较其他处理避免了单一氮源造成的局限性, 多样性指数增加, 菌群组成结构优化, 碳水化合物代谢功能注释相对丰度较高, 提高了秸秆降解效率, 为进一步开发秸秆还田促腐菌剂提供基础。
References
[1] MARSCHNER P, KANDELER E, MARSCHNER B. Structure and function of the soil microbial community in a long-term fertilizer experiment[J]. Soil Biology and Biochemistry, 2003, 35(3): 453–461
[2] PETERSSON L, KVIEN I, OKSMAN K. Structure and thermal properties of poly (lactic acid)/cellulose whiskers nanocomposite materials[J]. Composites Science and Technology, 2007, 67(11/12): 2535–2544
[3] YANG H Y, WU H, WANG X F, et al. Selection and characteristics of a switchgrass-colonizing microbial community to produce extracellular cellulases and xylanases[J]. Bioresource Technology, 2011, 102(3): 3546–3550
[4] WANG X J, YUAN X F, WANG H, et al. Characteristics and community diversity of a wheat straw-colonizing microbial community[J]. African Journal of Biotechnology, 2011, 10(40): 7853–7861
[5] ULRICH A, KLIMKE G, WIRTH S. Diversity and activity of cellulose-decomposing bacteria, isolated from a sandy and a loamy soil after long-term manure application[J]. Microbial Ecology, 2008, 55(3): 512–522
[6] LI P P, WANG X F, YUAN X F, et al. Screening of a composite microbial system and its characteristics of wheat straw degradation[J]. Agricultural Sciences in China, 2011, 10(10): 1586–1594
[7] FENG Y J, YU Y L, WANG X, et al. Degradation of raw corn stover powder (RCSP) by an enriched microbial consortium and its community structure[J]. Bioresource Technology, 2011, 102(2): 742–747
[8] QINGGEER, GAO J L, YU X F, et al. Screening of a microbial consortium with efficient corn stover degradation ability at low temperature[J]. Journal of Integrative Agriculture, 2016, 15(10): 2369–2379
[9] YU X F, BORJIGIN Q, GAO J L, et al. Exploration of the key microbes and composition stability of microbial consortium GF-20 with efficiently decomposes corn stover at low temperatures[J]. Journal of Integrative Agriculture, 2019, 18(8): 1893–1904
[10] PARKS D H, BEIKO R G. Identifying biologically relevant differences between metagenomic communities[J]. Bioinformatics, 2010, 26(6): 715–721
[11] NELSON C E, GARDNER J G. In-frame deletions allow functional characterization of complex cellulose degradation phenotypes in[J]. Applied and Environmental Microbiology, 2015, 81(17): 5968–5975
[12] MILLWARD-SADLER S J, DAVIDSON K, HAZLEWOOD G P, et al. Novel cellulose-binding domains, NodB homologues and conserved modular architecture in xylanases from the aerobic soil bacteriasubsp.and[J]. Biochemical Journal, 1995, 312(1): 39–48
[13] WU Y R, HE J Z. Characterization of a xylanase-producingstrain J3-8 and its genome analysis[J]. Scientific Reports, 2015, 5: 10521
[14] WANG W D, YAN L, CUI Z J, et al. Characterization of a microbialcapable of degrading lignocellulose[J]. Bioresource Technology, 2011, 102(19): 9321–9324
[15] DA X Y, JIANG F, CHANG X L, et al.sp. nov., isolated from soil in Antarctica[J]. International Journal of Systematic and Evolutionary Microbiology, 2015, 65(11): 3841–3846
[16] WON K H, KOOK M, YI T H.sp. nov., isolated from soil of a bamboo plantation[J]. Antonie van Leeuwenhoek, 2015, 107(2): 565–573
[17] KANG J Y, CHUN J, JAHNG K Y.sp. nov., isolated from freshwater, and emended description of the genus[J]. International Journal of Systematic and Evolutionary Microbiology, 2013, 63(5): 1633–1638
[18] 周丽沙, 李慧, 张颖, 等. 石油污染土壤鞘氨醇单胞菌遗传多样性16S rDNA-PCR-DGGE分析[J]. 土壤学报, 2011, 48(4): 804–812 ZHOU L S, LI H, ZHANG Y, et al. Analysis ofgenetic diversity in petroleum-contaminated soils by using PCR-DGGE technique[J]. Acta Pedologica Sinica, 2011, 48(4): 804–812
[19] DUAN J, LIANG J D, DU W J, et al. Biodegradation of kraft lignin by a bacterial strainsp. HY-H[J]. Advanced Materials Research, 2014, 955/959: 548–553
[20] XING W, LI J L, LI P, et al. Effects of residual organics in municipal wastewater on hydrogenotrophic denitrifying microbial communities[J]. Journal of Environmental Sciences, 2018, 65: 262–270
[21] LA SCOLA B, MALLET M N, GRIMONT P A D, et al.sp. nov.,sp. nov. andsp. nov., isolated from hospital water supplies, and emendation of the genus(Das1996)[J]. International Journal of Systematic and Evolutionary Microbiology, 2003, 53(1): 15–20
[22] XU L, ZHANG Y, READ N, et al.sp. nov., a novel species isolated from the roots ofin China[J]. Antonie van Leeuwenhoek, 2017, 110(11): 1475–1483
[23] HASSAN Y I, LEPP D, ZHOU T. Genome assemblies of three soil-associatedspecies:,.,and.[J]. Genome Announcements, 2015, 3(3): e00514-15
[24] JIN L, LEE H G, KIM H S, et al.sp. nov., a stalked bacterium isolated from a eutrophic reservoir[J]. International Journal of Systematic and Evolutionary Microbiology, 2013, 63(7): 2559–2564
[25] SUN L N, YANG E D, WEI J C, et al.sp. nov., a stalked bacterium isolated from rhizosphere soil[J]. International Journal of Systematic and Evolutionary Microbiology, 2015, 65(12): 4374–4380
[26] JACKSON C A, COUGER M B, PRABHAKARAN M, et al. Isolation and characterization ofsp. strain YS-1r that degrades lignin in plant biomass[J]. Journal of Applied Microbiology, 2017, 122(4): 940–952
[27] LAUS M C, VAN BRUSSEL A A N, KIJNE J W. Role of cellulose fibrils and exopolysaccharides ofin attachment to and infection ofroot hairs[J]. Molecular Plant-Microbe Interactions, 2005, 18(6): 533–538
[28] TER BEEK J, GUSKOV A, SLOTBOOM D J. Structural diversity of ABC transporters[J]. Journal of General Physiology, 2014, 143(4): 419–435
[29] UEKI A, KODAMA Y, KAKU N, et al.gen. nov., sp. nov., a facultatively anaerobic, fermentative stalked bacterium in the classisolated from rice plant roots[J]. The Journal of General and Applied Microbiology, 2010, 56(3): 193–203
[30] KODAMA Y, WATANABE K.sp. nov., a facultatively anaerobic, fermentative, prosthecate bacterium isolated from a cellulose-fed microbial fuel cell[J]. International Journal of Systematic and Evolutionary Microbiology, 2011, 61(8): 1781–1785
[31] ABRAHAM W R, ESTRELA A B, NIKITIN D I, et al.sp. nov.,sp. nov. andsp. nov., prosthecate bacteria from aquatic habitats[J]. International Journal of Systematic and Evolutionary Microbiology, 2010, 60(8): 1837–1843
[32] 司美茹, 赵云峰, 苏涛. 高效降解烷烃的无色杆菌XL株的分离鉴定及其降解特性[J]. 土壤通报, 2011, 42(3): 562–567 SI M R, ZHAO Y F, SU T. Isolation and identification of a high-efficiency alkane-degradingXL strain and its degradation characteristics[J]. Chinese Journal of Soil Science, 2011, 42(3): 562–567
[33] 唐玉斌, 孙常宇, 陈芳艳, 等. 一株[艹屈]高效降解菌的分离鉴定及其降解特性[J]. 微生物学通报, 2009, 36(4): 593–597 TANG Y B, SUN C Y, CHEN F Y, et al. Isolation and identification of a chrysene-degrading strain and its degradation characteristics[J]. Microbiology, 2009, 36(4): 593–597
[34] WAKARCHUK W W, BROCHU D, FOOTE S, et al. Proteomic analysis of the secretome ofATCC 484 andATCC 482[J]. PLoS One, 2016, 11(3): e0151186
[35] ZHUANG W P, ZHANG S Z, XIA X, et al. Draft genome sequence ofT26Tand comparative analysis of six[J]. Standards in Genomic Sciences, 2015, 10: 104
[36] KUPRYASHINA M A, PETROV S V, PONOMAREVA E G, et al. Ligninolytic activity of bacteria of the generaand[J]. Microbiology, 2015, 84(6): 791–795
[37] 肖建军, 李亚龙, 杨琦. 寡养单胞菌降解石油污染土壤中的甲苯[J]. 环境工程, 2018, 36(4): 186–189 XIAO J J, LI Y L, YANG Q. Degradation of toluene in petroleum contaminated soil bysp. MJ-1[J]. Environmental Engineering, 2018, 36(4): 186–189
[38] MADHAIYAN M, JIN T Y, ROY J J, et al.sp. nov., an endophytic N-fixing bacterium isolated from root tissue ofL.[J]. International Journal of Systematic and Evolutionary Microbiology, 2013, 63(7): 2477–2483
[39] LEWIN G R, JOHNSON A L, SOTO R D M, et al. Cellulose-enriched microbial communities from leaf-cutter ant () refuse dumps vary in taxonomic composition and degradation ability[J]. PLoS One, 2016, 11(3): e0151840
[40] ABT B, TESHIMA H, LUCAS S, et al. Complete genome sequence oftype strain (4M15T)[J]. Standards in Genomic Sciences, 2011, 4(1): 2–12
[41] ZIEMER C J. Newly cultured bacteria with broad diversity isolated from eight-week continuous culture enrichments of cow feces on complex polysaccharides[J]. Applied and Environmental Microbiology, 2014, 80(2): 574–585
[42] LIANG Y L, ZHANG Z, WU M, et al. Isolation, screening, and identification of cellulolytic bacteria from natural reserves in the subtropical region of China and optimization of cellulase production byME27–1[J]. Biomed Research International, 2014, 2014: 512497
Straw degradation ability and composition of microbial consortium for corn straw decomposition at low temperature*
Qinggeer1,2, YU Xiaofang1,2**, GAO Julin1,2**, WANG Zhigang1,2, HU Wanji1, Naoganchaolu2,3, WANG Zhen2,3, HU Shuping2,4, SUN Jiying1,2, QU Jiawei1,2
(1. Agricultural College, Inner Mongolia Agricultural University, Hohhot 010019, China; 2. Key Laboratory of Crop Cultivation and Genetic Improvement in Inner Mongolia Autonomous Region, Hohhot 010019, China; 3. Horticulture and Plant Protection College, Inner Mongolia Agricultural University, Hohhot 010019, China; 4. Vocational and Technical College, Inner Mongolia Agricultural University, Baotou 014100, China)
It is common practice to return field straw with urea to accelerate decomposition. To improve the microbial screening methodology and investigate the microbes responsible for decomposition of corn straw, nitrogen acclimatization of the microbial consortium GF-20 (GF-20) was performed. Compositional differences in the cultured microbial consortium and the microbe source were also evaluated. GF-20 was cultured until the 10thgeneration in variable nitrogen conditions [ammonium sulfate (N1), mixtures of ammonium sulfate and urea (N2-N5), and urea (N6)]. The corn straw decomposition ratio was determined to estimate the activity of the composite microbial system, and the composition diversity and function were analyzed by MiSeq high-throughput sequencing. The results showed that the N2 degradation rate was significantly higher than the other treatments. The bacterial source alpha diversity index (ADI) was significantly higher than the cultured microbial consortium, and the N2 ADI was significantly higher than the other nitrogen treatments. The bacterial composition also significantly differed between the source and consortium, as well as among the nitrogen treatments. The N2 treatment yielded the most diverse bacterial composition, with richer and more uniform flora structures and a higher carbohydrate metabolic pathway activity (which promotes corn straw degradation). Functional microbial strains involved in corn straw degradation were obtained after restrictive sub-generation of the microbial sources, which can accelerate corn straw degradation. The highest corn straw degradation efficiency of microbial consortium was observed with the 0.16% ammonium sulfate + 0.04% of urea nitrogen (N2) treatment. These findings provide a basis for developing microbial consortium used in commercial production decomposition.
Original microbe source; Microbial consortium; Nitrogen resources; High throughput sequencing; Composition diversity; Corn straw degradation
s: YU Xiaofang, E-mail: yuxiaofang75@163.com; GAO Julin, E-mail: nmgaojulin@163.com
S145.6
10.13930/j.cnki.cjea.200128
Feb. 26, 2020;
Jun. 21, 2020
青格尔,于晓芳, 高聚林, 王志刚, 胡万吉, 闹干朝鲁, 王振, 胡树平, 孙继颖, 屈佳伟. 玉米秸秆低温降解复合菌系降解能力及微生物组成研究[J]. 中国生态农业学报(中英文), 2020, 28(11): 1753-1765
Qinggeer, YU X F, GAO J L, WANG Z G, HU W J, Naoganchaolu, WANG Z, HU S P, SUN J Y, QU J W. Straw degradation ability and composition of microbial consortium for corn straw decomposition at low temperature[J]. Chinese Journal of Eco-Agriculture, 2020, 28(11): 1753-1765
* 国家自然科学基金项目(31760353)、内蒙古自然基金项目(2020MS03086, 2018ZD02)、国家重点研发计划项目(2017YFD0300804)、国家玉米产业技术体系项目(CARS-02-63)、农业部华北黄土高原地区作物栽培科学观测实验站(25204120)和内蒙古农业大学高层次人才引进科研启动项目(NDYB2016-15)资助
于晓芳, 主要从事作物生理生态研究, E-mail: yuxiaofang75@163.com; 高聚林, 主要从事作物生理生态研究, E-mail: nmgaojulin@163.com
青格尔, 主要从事玉米生理生态研究。E-mail: qinggeer001@163.com
2020-02-26
2020-06-21
* This study was supported by the National Natural Science Foundation of China (31760353), the Inner Mongolia Natural Sciences Foundation (2020MS03086, 2018ZD02), the National Key Research and Development Project of China (2017YFD0300804), the Earmarked Fund for China Agriculture Research System (CARS-02-63), the Crop Science Observation & Experiment Station in Loess Plateau of North China, Ministry of Agriculture, China (25204120) and the Advanced Talented Scholars of Inner Mongolia Agricultural University, China (NDYB2016-15).
