口-面-指综合征Ⅰ型1 例报告及文献回顾
2020-11-06暴丽莎
暴丽莎 栗 静 刘 芳
中国人民解放军白求恩国际和平医院新生儿科(河北石家庄 050082)
口-面-指(趾)综合征(orofaciodigital syndrome,OFD)是一种主要表现为口腔异常、面部和骨骼畸形的罕见遗传性疾病。OFD可依据临床表现分为13型,其中 OFD I 型(OMIM # 311200)呈X连锁显性遗传,在男性中胚胎时期具有致死性[1]。由于OFD I型极为少见,早期难以明确诊断。本文回顾分析1 例OFD I型新生儿的临床表现、基因检测结果及近1 年随访结果,并检索相关文献,探索OFD 1型的临床特点。
1 临床资料
患儿 女,2日龄,因吸吮差、先天性腭裂于新生儿科就诊。患儿系G2 P2,胎龄38+1周,其母因瘢痕子宫择期剖宫产,出生体质量3 100 g,身长51 cm,头围34 cm,无窒息抢救史。生后因易呛奶于当地就诊发现腭裂,转入中国人民解放军白求恩国际和平医院。患儿母亲28岁,父亲29岁,母孕期无特殊服药史及接触史;父母非近亲结婚,父母表型未见明显异常。患儿有一同胞哥哥,3岁,智力及体格发育无异常;患儿奶奶为先天性唇裂。入院体格检查:生命体征平稳,刺激反应好,哭声响亮,皮肤红润,眼距宽,小眼裂,鼻根宽,可见粟栗疹,内眦赘皮,腭裂,两叶舌,舌错构瘤,耳位低,杯状耳,右足3 趾短趾骨,左足多趾并1、2 趾间距宽(图1);女婴生殖器,外阴无异常;心肺腹无异常;四肢活动好,肌张力正常,新生儿反射正常存在。实验室检查:血糖4.2 mmol/L,三大常规、肝肾功能无异常;TORCH病原检测阴性。腹部超声示肝、胆、胰、脾、门静脉未见明显异常。双肾超声未见异常。心脏彩超示心内结构未见明显异常。颅脑磁共振平扫脑实质未见明显异常。液相串联质谱法分析血氨基酸和酰基肉碱谱无异常,尿有机酸综合分析未见显著异常。住院治疗7 天,患儿能自行吸吮、无呛咳,出院门诊随访。10月龄时,患儿运动发育基本同同龄儿,语言发育稍迟缓,无惊厥发生;15 月龄时,患儿运动基本同同龄儿,语言发育迟缓,能说单词,牙齿未萌出。

图1 患儿特殊体貌
患儿临床疑似OFD,为进一步明确诊断,经医学伦理审查批准,患者家属知情同意,对患儿及其父母进行全外显子测序和相关分析。采集患儿及其父母外周血,提取有核细胞基因组DNA。通过Illumina 公司NovaSeq 6000系列测序仪进行高通量测序(PE150),按Illumina 标准流程测序。使用Burrows-Wheeler Aligner软件测序数据与参考基因组GRCh37/hg19比对,使用GATK 软件分析出单核酸多态性、插入缺失标记;最后根据测序深度、突变质量对检测到的单核酸多态性插入缺失标记进行过滤和筛选,得到高质量可靠的变异。使用自主开发的变异注释软件对检测到的高质量变异进行各大数据库(dbSNP、千人基因组、ExAC、ESP等频率数据库及OMIM、HGMD、ClinVar等)的关联注释。借助Provean、SIFT、Polyphen 2-HVAR、Polyphen2-HDIV、M-Cap、Revel、Mutationtaster等蛋白结构预测软件,MaxEntScan剪切位点预测软件等对其危害性进行分析,筛选出对蛋白结构可能有有害影响的变异。结果发现,患儿X 染色体OFD 1基因chrX:13754799 杂合变异,c.312+2(IVS 3)T>A,即位于X染色体13754799基因的第3个内含子第2位碱基T突变为A,距离第1外显子边界(c.312)2 bp的位置;患儿父母均未检出该变异(图2)。考虑患儿为杂合新发变异,符合X 染色体显性遗传疾病发病机制。多个剪接位点危害性软件预测,该变异位点会影响外显子的剪切,MaxEntScan:Deleterious(9.06-> 0.88),dbscSNV:Deleterious(1.0000|0.9260)。根据美国医学遗传学与基因组学学会(American College of Medical Geneticsand Genomics,ACMG)指南判定该位点变异性质为致病。查阅正常人群携带率数据库、EXAC 东亚人数据库未见收录。患儿最终诊断为OFD 1基因新发变异致OFD I 型。系统通过自有算法对变异数据进行分析,提交可能的拷贝数变异(copy number variation,CNV)结果显示未发现临床明确致病的CNV变异。
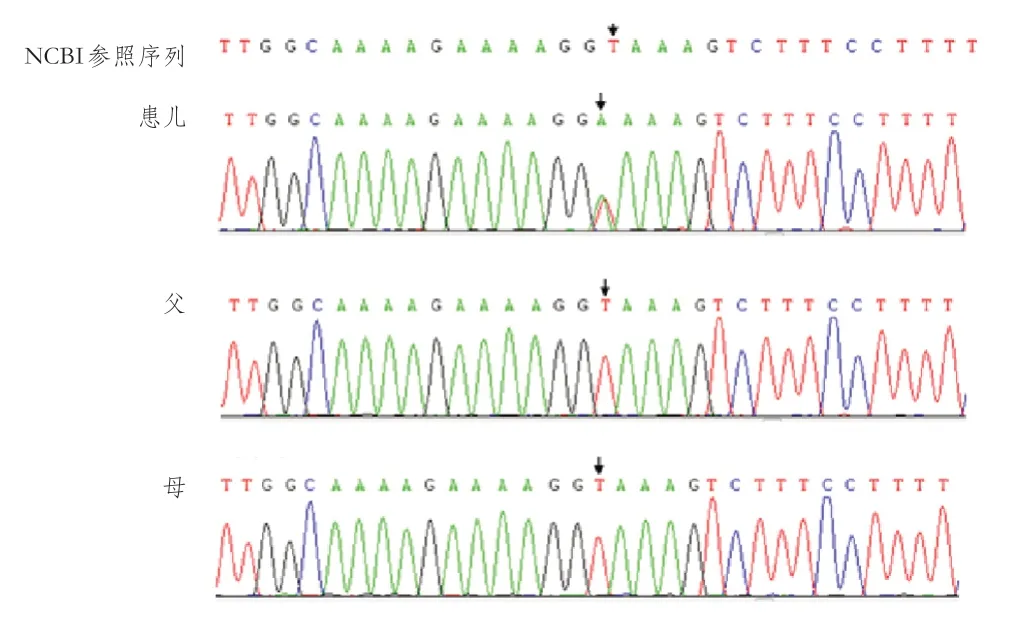
图2 患儿及父母OFD1 基因测序图
2 讨论
OFD是一种以颅面、口腔和指(趾)骨缺陷,以及脑和肾发育异常为表现的综合征。1954 年首次报道8例女性患者,并推测其可能为一种X连锁显性遗传性疾病。根据临床表现,OFD可分为13个类型,其中以I型最为常见[2-3]。OFD I型患者通常有比较典型的临床特征:①颅面部,主要异常包括面部不对称、额突、眼距宽、宽鼻根及面部粟栗疹;②口腔畸形,特征性表现为“裂”与“分叶”,包括上唇正中裂、腭裂、分叶舌(约30%分为2 叶、45%分为3 叶或更多)、口腔系带增生、舌错构瘤(75%)、牙槽嵴增厚及牙列异常、唇缘异常、牙釉质发育异常、小下颌等;③骨骼方面,约50%~70%患者存在指(趾)畸形,包括并指(趾)、指(趾)过短或弯曲,有时表现为单侧指(趾)畸形,另有文献报道约88%OFD患者指(趾)畸形中无多指(趾)改变[4];④其他,约60%的OFD 1型有中枢神经系统异常,如脑结构发育异常,发育迟缓和智力障碍等[5];此外,有研究认为多囊肾是OFD I型区别于其他分型的主要特征之一,但多数发病年龄在18岁以后,在儿童期没有多囊肾的表现[6-9]。本例患儿有突出的特征性表现:①颅面部发育不对称,额突,眼距宽,内眦赘皮,小下颌;②腭裂,分叶舌(2叶),舌错构瘤,牙槽嵴增厚,唇缘无明显异常;③右足3趾短趾骨,左足多趾并1、2趾间距宽。患儿表型完全符合OFD I型的外观特征。文献报告,约60%的ODF 1型患者可能出现神经系统异常,包括脑积水、胼胝体发育不良、精神发育迟滞等情况[10]。本例患儿目前已随访至1岁3个月,颅脑磁共振未见实质性病变,胼胝体发育无异常,无脑积水发生。另患儿双肾超声未见异常,肾功能无异常;大运动发育无异常,神经精神发育基本同同龄儿,但语言发育稍落后于同龄儿,能说单词,但吐字不清,考虑可能与患儿腭裂及分叶舌有关。患儿至1 岁3 个月仍无乳牙萌出;口腔X 线摄片检查显示牙胚存在,考虑为乳牙萌出延迟,与口腔畸形有关。此外,有文献报道OFD 1型患者除多囊肾外,还可能发展为肝脏和胰腺纤维囊性疾病[3]。建议对OFD 1型患者进行长期追踪随访,对内脏的发育进行常规监测。
OFD I 型是由位于 Xp 22.2 的OFD 1基因变异引起X 连锁显性遗传病[2,7],发病率为1:50000 至1:250000[11-12]。OFD 1基因有23 个编码外显子,编码由1 011个氨基酸组成的OFD1蛋白[13-16]。迄今,已在OFD 1中鉴定出172 种不同的变异。目前关于OFD 1基因变异的位置已扩展到23 个编码外显子中的前17 个外显子。已报道的OFD1基因变异大多位于外显子3、7、8、9、12、13和16,这些外显子可能代表变异热点[17]。本例患儿全外显子基因组测序及生物信息分析显示其X染色体13754799基因第3个内含子有一处杂合变异位点c.312+2(IVS3)T>A,属于剪切变异,导致基因功能可能丧失。ACMG变异分级为致病。查询HGMDpro数据库,c.312+2(IVS3)T>A变异在国内外尚未见报道。
目前有一些关于OFD1基因型与表型相关性的研究[18]。如有报道一家系OFD1基因第16外显子2个碱基对缺失引起移码变异,导致蛋白表达过早终止,引起蛋白功能丧失;在这一家系中,母亲表现出比其女儿更轻的表型,可见OFD I型的临床特征与变异类型之间没有关联[12]。OFD I型基因变异位置与某些临床特征具有相关性,如有研究指出,脑部结构异常可能与外显子7和16的变异有关,肾囊肿可能与外显子3、9、12变异相关,而外显子3、8、9、13和16的变异可能与智力障碍具有相关性[5]。OFD 1基因的临床变异性对准确诊断和遗传咨询提出了挑战,需要引起重视。
OFD I型诊断主要依据典型的临床表现,如女性患者具有典型的颅面部、口腔和指(趾)畸形,成人多伴有多囊肾,甚至可能有5%~15%伴有肾衰竭[19],结合分子遗传学检查可确定诊断。国外文献报道,胎儿时期超声检查发现胼胝体缺如,经绒毛膜取样行基因检测和染色体检测可确诊OFD I型。
目前OFD I型尚无特效治疗方法,主要是对症治疗。对于面部畸形或口腔畸形可予美容整形矫治。对语言发育迟缓、构音障碍可整形后进行语言训练治疗。定期检查神经系统发育情况、颅脑及肾脏有无病变。因OFD I型属于X染色体显性遗传,建议母亲再次妊娠时做羊水穿刺行基因检测,以有效避免此类缺陷患儿出生。
综上,临床对于特征性面部、口腔和手指(趾)畸形的患儿应考虑到OFD I型的可能,尽早进行遗传咨询、基因筛查,以明确诊断。
