GPI 基因突变致葡萄糖磷酸异构酶缺乏症1 例报告并文献复习
2020-11-06汪爱平刘玲娟肖阳阳毛定安刘利群
黄 鹏 唐 黎 汪爱平 刘玲娟 熊 洁 肖阳阳 毛定安 刘利群
中南大学湘雅二医院1.儿科,2.儿童脑发育与脑损伤研究室(湖南长沙 410011)
葡萄糖磷酸异构酶(glucose-6-phosphate isomerase,GPI)缺乏症(OMIM:172400)是一种罕见的常染色体隐性遗传病,发病率低[1],1968年首次报道[2],其主要临床表现为慢性非球形大细胞性溶血性贫血,严重者可有神经肌肉功能障碍、胎儿水肿,可致新生儿死亡[3-4]。目前GPI 缺乏症报道较少,临床诊断较为困难。本文回顾分析1例GPI 缺乏症患儿的临床资料和基因检测结果,并结合文献进行总结。
1 临床资料
患儿,男,4岁3月龄。因反复皮肤黄染、贫血4年余,皮疹4天,发热2天入院。患儿入院前4天被蚊虫叮咬后双下肢出现皮疹并破溃,入院前2 天接种麻腮疫苗后发热,面色苍白,皮肤黄染加重。患儿为G1P1,足月顺产,出生体质量4.3 kg,血型为A型、RH(D)阳性。生后6小时出现黄疸、重度贫血,治疗后好转(具体不详)。此后贫血、皮肤黄染反复发生,伴有肝脾肿大。每隔1~3 个月输注红细胞治疗,4 岁时因巨脾行脾切除术。患儿1岁才能独坐,2岁扶站,3岁能独走,易摔跤;能正常言语交流,理解能力可,反应敏捷,能自行穿衣、洗漱。父亲体健,母亲患有地中海贫血,母亲家族有地中海贫血史(具体不详)。
入院体格检查:体温37.4℃,脉搏130次/min,呼吸32次/min,身高97 cm(<-1SD),体质量14.5 kg(<-1 SD),神清,巩膜黄染,眼睑、唇黏膜、甲床苍白;双小腿内侧皮肤可见散在丘疹,丘疹中心可见破溃及黄色分泌物,周围有红晕;咽部充血,双侧扁桃体III度;腹部膨隆,肝右肋缘下8 cm,剑突下7 cm,质韧,无压痛;脊柱、四肢无畸形,双下肢肌张力稍低,四肢肌力正常;行走时步伐较小、速度较慢,独自行走约30 米后诉双侧膝关节疼痛;膝反射、跟腱反射可引出,病理征、脑膜刺激征阴性。实验室检查:血常规白细胞计数(WBC)28.38×109/L,血红蛋白(Hb)63 g/L,平均红细胞体积(MCV)119.8 fL,网织红细胞0.62%;外周血涂片示成熟红细胞大小不均,可见大量嗜碱性点彩红细胞、嗜多色性红细胞;谷氨酸氨基转移酶69.2 U/L,天冬氨酸氨基转移酶269.5 U/L,总胆红素77.5 μmol/ L,间接胆红素36.2 μmol/L,乳酸脱氢酶698.4 U/L;铁蛋白 31 155.2 ng/mL;超敏C反应蛋白35.78 mg/L,降钙素原2.4 ng/L;肌酶、病毒性肝炎抗原抗体、自身免疫性肝炎抗体、狼疮抗体谱、抗核抗体谱、血管炎抗体谱、抗O抗体、类风湿因子未见异常;结核斑点试验、结核杆菌抗体、巨细胞病毒DNA、EB病毒DNA、血培养均阴性;血尿代谢筛查、血清铁、铁染色、葡萄糖-6-磷酸脱氢酶(G6PD)酶、丙酮酸激酶、血红蛋白电泳、直接抗人球蛋白试验、红细胞脆性试验、酸溶血试验、骨髓细胞学检查、染色体检查均未见异常。髋膝关节X线、头颅及脊髓MRI、四肢肌电图无异常;腹部CT 示肝大;膝关节MRI 示双膝关节少量积液并滑膜增厚。
根据患儿临床表现及辅助检查结果,无法用常见的溶血性疾病解释,怀疑遗传代谢性疾病可能,经医院伦理审核及家属知情同意后,对患儿及父母行全外显子检测及Sanger 测序验证。结果发现患儿GPI基因第6 号外显子存在c.553 T>A 纯合错义变异(NM_001289790,F 185 I),分别遗传自父母,见图1。查阅千人基因组计划、ClinVar数据库、人类基因突变数据库,均未见该位点变异报道。根据美国医学遗传学与基因组学学会(American College Medical Genetics and Genomics,ACMG)指南,该基因变异的致病性分析为III类,即临床意义未明。该变异位点位于GPI蛋白第一个结构域内,经Polyphen 2 软件预测该变异极可能致病(致病性相关分数为0.983);Provean 软件预测该变异为影响蛋白功能的有害变异(影响分数为-4.995)。构建基因变异前后GPI 蛋白结构图,可见蛋白空间构象发生改变(图2)。对GPI基因进行蛋白质二级结构预测显示,该基因编码的蛋白质中,包含24个α-螺旋、8个β-折叠、33个无规卷曲,其中二级结构占比为α-螺旋42.64%、β-折叠8.49%、无规卷曲48.87%,c.553T>A(p.Phe185Ile)变异位于卷曲结构内,为错义变异,位于蛋白编码区,推测该变异可能导致功能上的改变。同时结合患儿临床表现、血生化检查结果和家系特点,可判定该突变为致病突变。
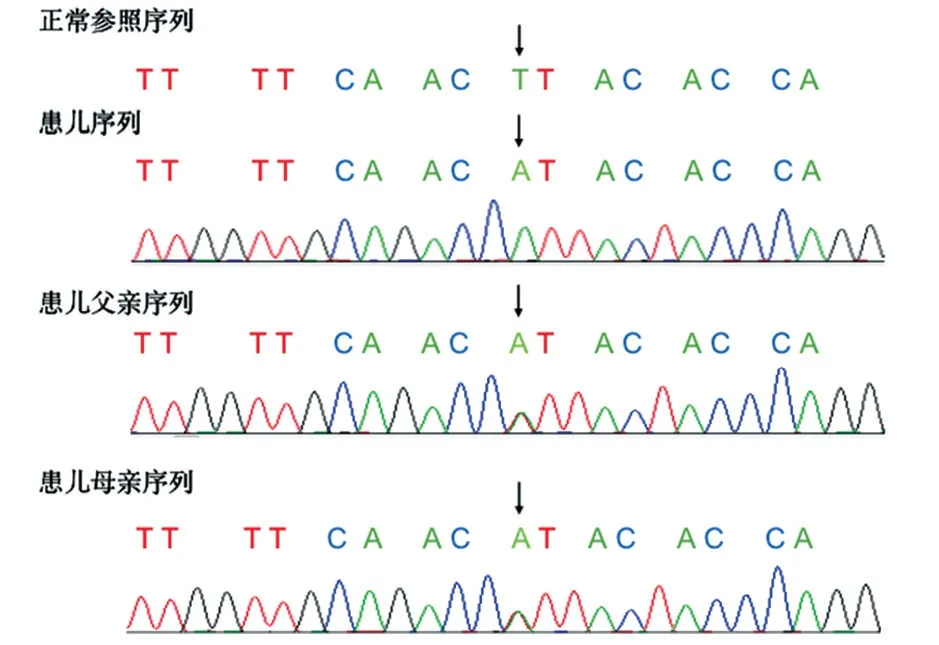
图1 患儿及父母GPI 基因测序图

图2 GPI 蛋白结构预测图
结合临床表现、基因检查结果以及致病基因遗传方式,患儿确诊为GPI缺乏症。入院后予积极抗感染、输注浓缩红细胞、护肝等对症支持治疗,贫血、黄疸明显改善。出院后随访1 年,患儿病情稳定,Hb 维持在70~80 g/L,期间只输4次红细胞,未再接种疫苗。目前服用去铁酮片。随访过程中Gesell发育量表评估示大运动88分,精细运动94分,适应性92分,语言90分,个人社交87分。
2 讨论
GPI缺乏症是由于编码GPI蛋白的GPI基因发生变异所致[2]。GPI基因位于19q13.1上,包含18个外显子、长约1.9 kb的cDNA,编码成含有558个氨基酸的GPI蛋白,该蛋白是一种同型二聚体酶,在所有组织中均有表达,主要参与糖酵解第二步骤[5]。
以“葡萄糖-6-磷酸异构酶缺乏症”、“葡萄糖磷酸异构酶缺乏症”为关键词和主题词精确检索中国期刊全文数据库(CNKI)及万方数据库,以“Glucose-6-phosphate isomerase deficiency”、“Glucose phosphate isomerase deficiency”为关键词和主题词检索PubMed数据库,均从建库至2020年4月。共检索到269篇文献,其中临床资料齐全、经基因检测确诊的英文文献31篇、中文文献1篇,排除重复报道的病例,共有患者69例。其中男36 例、女33 例,以欧美、印度人居多,多在出生或婴幼儿期起病。患者主要临床表现为反复面色苍黄、皮肤黄染、贫血貌,此外还可伴肝脾大、胆囊结石、高胆红素血症、高网织红细胞血症、高铁蛋白血症,胆红素升高以间接胆红素为主,部分可有乳酸脱氢酶升高[3]。患者易反复发生感染,而感染、药物、疫苗接种可诱发严重的溶血危象[2,6]。部分患者可出现智能运动发育迟缓、肌张力降低、肌无力、面瘫、顽固性癫痫、共济失调、胎儿水肿及新生儿死亡[7-8]。其中有2例GPI缺乏症患者合并G6PD缺乏症[3,9],推测G6PD缺乏可能增强GPI活性,并可改善GPI缺乏后的生化结果及临床后果[9]。本例患儿自出生起反复出现黄疸、贫血、肝脾肿大、肌张力稍低,实验室检查显示大细胞低色素性贫血、高间接胆红素血症、高网织红细胞血症、高铁蛋白血症、高乳酸脱氢酶血症,与既往文献报道相符合。
GPI缺乏症临床表现轻重不一,轻度患者可存活至成年,严重者出生即可死亡。但GPI 缺乏的程度与贫血、黄疸的严重程度无关,且临床表型与基因型的异质性很大,一些纯合变异患者可能没有任何症状,而另一部分患者可能并发严重的神经肌肉功能障碍,且同一位点纯合变异导致的贫血与黄疸程度、输血依赖度也可能不同[2,5]。GPI缺乏症患者常规溶血性贫血筛查往往无异常,其诊断基础是GPI 活性测定,但有部分患者酶活性变化不显著,且GPI活性易受到输血等因素的影响。基因检测是目前确诊该病最精准的方法[10]。文献检索到的69例患者中复合杂合变异25例、纯合变异44例,涉及变异位点47个。变异类型包括错义变异、无义变异、移码变异及剪切位点变异,以错义变异最常见。变异位点以exon12处的c.1040G>A最多,该变异在合并神经肌肉损伤的印度GPI缺乏症患者中发生率较高。本例患儿具有典型的临床症状,有定期输血史,多次常规溶血性贫血筛查未见异常,考虑到反复多次输血可能影响GPI 的活性及实验室条件限制,未检测GPI 活性。基因检测证实本例患儿GPI基因c.553T>A(F185I)点突变,未见报道,经验证为致病性变异。
与国内报道的另1例GPI缺乏症不同的是[11],本例患儿发病年龄更早、溶血程度稍轻、在婴幼儿期出现大运动发育落后、出现新的基因变异,同时本例患儿双下肢肌张力稍低,行走后诉双膝关节疼痛,髋膝X 线不支持骨折,头颅及脊髓MRI、肌电图检查不支持神经肌肉病变。血常规中白细胞计数升高,以中性粒细胞为主,炎症指标升高,但膝关节局部无红肿、压痛、摩擦感,抗感染治疗后白细胞计数、炎症指标下降均不支持局部感染。结合患儿免疫、结缔组织、结核检查阴性及膝关节MRI检查结果,考虑上述症状由双膝关节滑膜炎所致可能性大,复习相关文献未见GPI缺乏症合并关节滑膜炎的报道。
目前GPI缺乏症无特异性治疗,患者常需依赖输血维持Hb水平稳定,其他治疗方法包括脾切除、血浆置换、祛铁治疗[5]。部分患儿脾切除可减轻贫血、降低输血需求[12]。由于感染、疫苗接种可能诱发溶血危象,故对于确诊患儿应尽量预防和控制感染,同时还需慎重考虑接种疫苗的时机和种类。本例患儿定期输红细胞,脾切除后贫血、黄疸较前减轻,输血频率由1~2个月1次延长至3~4个月1次,本次入院是由于感染、接种疫苗致使溶血加重,积极抗感染后病情好转。
综上所述,GPI缺乏症具有典型溶血性贫血的临床表现,易和其他溶血性疾病发生混淆。GPI 缺乏症在临床中罕见,临床医师对该疾病的认识较少,易致漏诊、误诊。因此,临床上对于反复发生溶血性贫血且常见病因无法解释时,应尽早行GPI 酶活性、基因检测帮助尽早诊断、干预。本研究发现一个未见报道的GPI基因突变,丰富了基因突变数据库,为今后该疾病的研究提供参考。
