如何提高脱碳液中总钒测定的准确度
2020-11-06张应应国家能源集团宁夏煤业有限责任公司煤制油化工质检计量中心宁夏银川750411
张应应(国家能源集团宁夏煤业有限责任公司煤制油化工质检计量中心,宁夏 银川 750411)
0 引言
费托合成脱碳单元是将来自费托合成单元的合成尾气首先通过水洗塔脱除尾气中的烃类物质,然后进入吸收塔由贫液、半贫液(统称为脱碳液)洗涤吸收CO2,脱除CO2后的脱碳净化气一部分返回到费托继续参与费托反应,另一部分送至低温油洗装置。吸收塔底部生成的KHCO3溶液首先进入富液闪蒸槽释放出部分夹杂在溶液中的CO和H2,闪蒸后的溶液进入加压再生塔通过加热再生产生脱碳液循环再利用。该脱碳液是以热碳酸钾溶液为主体,通过加入缓蚀剂五氧化二钒,在设备、管道表面形成一层钝化膜,起到防腐缓蚀的作用。由于装置内的还原性物质会将五价钒还原成低价态的钒,装置内的五价钒、总钒的含量能很好的反映设备腐蚀情况,所以总钒分析结果的准确性至关重要。
目前分析脱碳液中总钒采用的是氧化还原电位滴定法[1],原理是脱碳液在酸性介质中首先用H2O2破坏各种有干扰作用的还原性物质,并且使溶液脱色,过量的H2O2通过加热分解,然后用高锰酸钾将低价钒全部氧化为五价钒,过量的高锰酸钾用草酸还原,最后用硫酸亚铁铵滴定五价钒,根据硫酸亚铁铵消耗量来计算总钒含量。但样品经过前处理后,总钒的数据有时会出现倒挂现象,即总钒含量反倒低于五价钒的含量,有悖于常理。
文章研究了手动滴定法和电位滴定法的分析比对,分析问题产生的原因,是仪器测定环节还是样品前处理过程存在问题,并建立ICP直接测定法[2]对结果测定的准确性进行评判。
1 试验部分
1.1 试验仪器
(1) 916电位滴定仪:铂-甘汞氧化还原复合电极、螺旋搅拌器、20mL高精度滴定管单元。
(2) DB-3不锈钢调温电热板:温度范围:室温-300℃,控温精度:±1℃;功率:2000W,功率可调。
(3) 5100 ICP-OES 高分辨率等离子体发射光谱仪:可拆式矩管、雾化系统、甭管、空压机。
(4) 电子天平:MSA623S-1CE-DE最大称量620g,最大误差:0.025mg。
1.2 玻璃器皿
(1) 烧杯:100mL、250mL。
(2) 棕色试剂瓶:1L。
(3) 容量瓶:250mL、500mL。
(4) 移液管:5mL、10mL。
1.3 试验试剂
(1) 混酸:H2O:H2SO4:H3PO4=35:35:30 体积比配制。
(2) 硫酸溶液:1:1。
(3) H2O2:30%水溶液。
(4) 高锰酸钾溶液:2.5%。
(5) 草酸溶液:1%。
(6) 硫酸锰溶液:8%,新鲜配制的为略显肉红色,少许沉淀,放置时间较长时,生成沉淀较多,可取上层清液使用。
(7) 苯代邻氨基苯甲酸溶液(0.2%):0.2g苯代邻氨基苯甲酸溶解于含有 0.2g Na2CO3的100mL水中煮沸。
(8) 硫酸亚铁铵标准溶液:0.02mol/L。
(9) 钒标准溶液:1mg/mL。
(10) 硝酸溶液:1%(φ/%),用优级纯浓硝酸稀释配制。
1.4 试验方法
脱碳液用H2O2消除干扰物质脱色后,用高锰酸钾将低价钒氧化成高价钒,过量的高锰酸钾用草酸还原后分别用电位滴定法和手动滴定法进行方法比对,研究影响测定结果的因素,同时用ICP 光谱仪直接测定脱碳液的总钒。
2 结果与讨论
2.1 不同滴定方式的比对试验
参照企标《Q/SNZJ-004-J-2017—0056 碳酸钾脱碳液中总钒、五价钒的测定》[3]中的操作步骤,分别对贫液、半贫液、分两批次配制的25.00g/L钒标准溶液4种试样分别采用电位滴定法和用苯代邻氨基苯甲酸作指示剂手动滴定法进行试验比对,通过试验发现电位滴定法和手动滴定法两种方法结果基本一致,均会出现总钒测定结果低于五价钒的数据倒挂现象,说明问题不在于仪器测定环节,而在前处理过程。
进一步分析,前处理中双氧水加入的作用是破坏消除还原性物质的干扰,过量后经加热分解,而高锰酸钾的作用是将低价态钒氧化成五价钒,试验中为保证高锰酸钾的过量,溶液由蓝绿色变成肉红色,且保证2~3分钟不退色,充分说明高锰酸钾保证是过量的,所以造成总钒结果偏低就锁定在草酸的加入量这个因素,因为草酸本身是一种还原剂,过量的草酸会将高价态的钒又转化成低价态钒,进而对测定结果造成影响,所以草酸加入量是制约结果准确与否的一个至关重要的影响因素。
2.2 草酸加入量对测定结果的影响
参照企标《Q/SNZJ-004-J-2017—0056 碳酸钾脱碳液中总钒、五价钒的测定》中的操作步骤,通过改变草酸的加入量,加入量分别为刚褪色、过量1d、过量2d、过量5d、过量1mL、过量2mL,用916电位滴定仪测定25.00g/L钒标准溶液中总钒。通过试验可以得出:草酸的加入量对总钒测定结果有很大影响,随着草酸过量量的增大总钒测定值逐渐降低,草酸过量控制在1~2滴时更接近总钒的真实值,为保证草酸与过量的高锰酸钾充分反应,应逐滴缓慢加入,在滴加过程中不断摇动烧杯来加速反应,且每滴加一滴等待至少15s仔细观察颜色变化。
进一步分析,试验中草酸的加入量是根据颜色肉眼观察来实现的,要求待红色恰消失后过量1~2滴,即由橙红色变为黄色,颜色不易观察,且脱碳液样品自身带有颜色,颜色突变不明显,进而阻滞了颜色的判断,那么草酸加入量的准确性有待考量,所以目前最佳的方案是寻求一种直接测定法,避免前处理对总钒测定结果的影响。
2.3 ICP光谱仪直接测定法
2.3.1 方法原理
待测样品经酸酸化后,进入等离子发射光谱仪的雾化器中被雾化,由氩载气带入等离子体火炬中,目标元素在等离子体火炬中被气化、电离、激发并辐射出特征谱线,特征光谱的强度与试样中待测元素含量在一定范围内成正比。
2.3.2 仪器参考测量条件
不同型号的仪器最佳测试条件不同,根据仪器说明书条件要求优化测试条件。5100 ICO-OES仪器参考测量条件见表1。

表1 仪器参考测量条件
2.3.3 校准曲线的绘制
用1%硝酸依次配制0、20mg/L、40mg/L、60mg/L、80mg/L、100mg/L系列浓度的钒标准溶液,按照仪器参考测量条件调试仪器,将标准溶液由低浓度到高浓度依次导入ICP等离子发射光谱仪测量对应的发射强度,以钒质量浓度为横坐标,发射强度值为纵坐标,建立钒元素的校准曲线。通过条件选择试验得知波长在297.401nm下强度最大,线性最好,相关系数为0.9998,曲线可用。
2.3.4 测定
(1)钒标准溶液的测定。配制2%总钒标准溶液,测定该溶液密度为:1.2537g/cm3,得出钒标准溶液浓度为25.07g/L,将标准溶液稀释1000倍后,在与建立校准曲线相同的条件下用ICP光谱仪对稀释液进行测定,仪器显示钒(以V元素计)测定结果。
(2)试样的测定[4]。分别取3批次贫液、半贫液共计6个样品,进行适当稀释后,用与钒标准溶液测定相同的操作步骤进行试样精密度试验。
2.3.5 结果计算
脱碳液中总钒以质量浓度w计,数值以克每升(g/L)计表示,按式(1)计算:
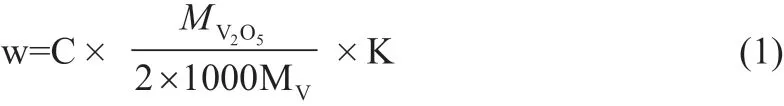
式中:w为脱碳液中总钒质量浓度,以V2O5/2计,单位为克每升(g/L);C为仪器显示的脱碳液中总钒的质量浓度,以V元素计,单位为毫克每升(mg/L);MV2O5为V2O5摩尔质量(181.88g/mol);MV为V摩尔质量(50.94g/mol);K为稀释倍数。
2.3.6 方法性能评价
试验参照HJ 168《环境监测 分析方法标准制修订技术导则》[5],对先后配制的两批次2%总钒标准溶液进行准确度试验,相对误差均小于2%;并对脱碳液中总钒的重复性进行确认,依次重复测定贫液、半贫液不同批次样品测定6次,测得的相对标准偏差见表5,可以看出,本试验条件下不同批次样品的相对标准偏差均小于2%,重复性试验数据均符合GB/T 27404—2008《试验室质量控制规范 食品理化检测》[6]附录中关于检测方法确认的技术要求。证明脱碳液中的总钒是可以通过ICP法直接测定的。
3 结语
文章首先通过手动滴定法与916电位滴定法两种方法比对,推断出造成总钒数据倒挂是由于样品前处理过程控制不当造成的,最终锁定问题在于草酸加入量对结果有直接的影响,通过改变草酸的加入量,得出草酸加入量为红色恰消失后过量1~2 滴,要求在滴加草酸时要缓慢,且充分摇动烧杯。为避免前处理对总钒测定结果的影响,建立了ICP光谱直接测定法,准确度试验相对误差<2%,精密度试验相对标准偏差控制5%范围内,达到标准GB/T 27404—2008《试验室质量控制规范 食品理化检测》中对精密度与准确度的要求,实现了脱碳液中总钒的准确分析。
