加速溶剂萃取-磁固相萃取-高效液相色谱-串联质谱法测定水产品中10种氟喹诺酮类药物残留
2020-11-02魏丹,国明,张菊
魏 丹, 国 明, 张 菊
(1. 河北经贸大学生物科学与工程学院, 河北 石家庄 050000; 2. 浙江省化工研究院有限公司分析测试中心, 浙江 杭州 310000)
氟喹诺酮类药物(fluoroquinolones, FQs)作为一类人工合成的抗菌药,具有广谱、高效、杀菌能力强、低毒等特点,已广泛用于水产、畜牧养殖业中动物疾病的预防和治疗。由于氟喹诺酮药物在养殖过程中的不合理使用和滥用,在食品中残留累积量呈递增趋势,对人类的健康造成潜在的风险。我国规定动物肌肉组织中氟喹诺酮类药物最大残留限量为10~500 μg/kg[1];自2015年,我国禁止在食用动物中使用培氟沙星、洛美沙星、诺氟沙星和氧氟沙星4类氟喹诺酮类抗生素。因此,建立水产品多种氟喹诺酮类药物残留的可靠检测方法,对保证动物源性食品安全具有积极意义。
目前,氟喹诺酮类药物残留的检测方法主要有毛细管电泳法(capillary electrophoresis, CE)[2,3]、高效液相色谱法(high performance liquid chromatography, HPLC)[4,5]和高效液相色谱-质谱法(high performance liquid chromatography-mass spectrometry, HPLC-MS)[6,7]等。HPLC和HPLC-MS在氟喹诺酮残留检测中广泛应用;其中,HPLC-MS结合了液相色谱的高分离能力和质谱高分辨率的检测能力,具有准确度高、灵敏度高、特异性和选择性强的特点,适用于喹诺酮类药物同时快速测定[8]。目前用于HPLC-MS/MS的样品前处理方法主要有加速溶剂萃取(accelerated solvent extraction, ASE)[9]、固相萃取(solid phase extraction, SPE)[10]、QuEChERS[11]和磁固相萃取(magnetic solid phase extraction, MSPE)[12]等。QuEChERS虽然操作简便,分析速度快,但是由于缺少了吸附洗脱这一过程,其净化效果弱于SPE。传统的SPE操作较为繁琐,MSPE作为一种新型SPE,具有操作简便、分离时间短,有机溶剂用量少等优点。然而,MSPE难以对固体样品中的目标分析物进行直接萃取。ASE具有溶剂消耗少、操作自动化程度高、提取充分、速度快等优点,适用于固体样品中的有机污染物的检测中[13],但其净化效果较差。
鉴于上述问题,本研究将ASE与MSPE相结合,建立了ASE-MSPE-HPLC-MS/MS同时检测水产品中10种氟喹诺酮类药物残留的分析方法,与单纯的ASE相比较,净化效果更为显著;与目前常用的SPE和QuECHERS净化方法相比较,ASE萃取液通过MSPE净化,保留了ASE和MSPE的优点,具有操作简便快速、有机溶剂用量少且不需要置换萃取溶剂等优点。该方法提取充分,净化效果良好,有效解决了固体样品基质效应导致的回收率低的问题,可以满足水产品中氟喹诺酮类药物残留检测的要求。
1 实验部分
1.1 仪器、试剂与材料
Agilent 1290 infinity高效液相色谱-6460 TripleQuad质谱仪(带自动进样器,美国Agilent公司); ZORBAX EclipsePlus C18(100 mm×3.0 mm, 1.8 μm,美国Agilent公司); Milli-Q超纯水器(美国Millipore公司); 0.22 μm微孔滤膜(上海安谱科学仪器有限公司); KQ-100DE超声波清洗器(昆山市超声仪器有限公司); SevenEasyS30型pH计(瑞士梅特勒-托利多公司); S-4700型场发射扫描电子显微镜(SEM)(日本Hitachi公司);傅里叶变换红外光谱分析仪(FTIR)(德国Bruker公司)。
10种氟喹诺酮标准物质:沙拉沙星(sarafloxacin, SAR);氧氟沙星(ofloxacin, OFL);恩诺沙星(enrofloxacin, ENR);丹氟沙星(danofloxacin, DAN);洛美沙星(lomefloxacin, LOM);培氟沙星(pefloxacin, PEF);环丙沙星(ciprofloxacin, CIP);依诺沙星(enoxacin, ENO);诺氟沙星(norfloxacin, NOR);双氟沙星(difloxacin, DIF);购于德国Dr. Ehrenstorfer公司。甲醇、乙腈均为色谱纯(美国Sigma-Aldrich公司);二氯甲烷、乙酸、氨水、乙酸铵等为分析纯,购自杭州双林化工试剂厂;实验用水为超纯水(电阻率≥18.2 MΩ·cm),由Millipore超纯水发生器产生。
纳米零价铁(nZVI,纯度>99.5%)(深圳微纳科技有限公司);氧化石墨烯(GO,纯度>99%)(南京先丰纳米材料科技有限公司)。
黄鱼、草鱼、黑鱼、明虾和沼虾为本地市售。
1.2 溶液配制
准确称取10种氟喹诺酮各0.100 0 g于100 mL容量瓶中,用甲醇溶解并定容,得到1 g/L标准储备溶液,然后存于4 ℃冰箱内避光保存。使用时,以空白基质提取液稀释成系列浓度的混合基质匹配标准工作溶液,现用现配。
1.3 磁性净化材料的制备及表征
磁性复合材料(GO@nZVI)的制备采用室温溶剂自组装法[14]。首先配制GO储备液:准确称取适量的GO,在水中超声分散15 min,配制成1 g/L的GO储备液。在25 ℃下将1.000 g nZVI加入10 mL去离子水中,超声15 min。然后加入50 mL的GO储备液,涡旋振荡30 s。最终生成黑色沉淀,使用磁铁分离净化,上清液为澄清液体,收集的沉淀物为GO@nZVI磁性复合材料,作为MSPE净化材料。
1.4 样品前处理
1.4.1样品采集及处理
将鱼去皮、去骨、去鳞,取肌肉组织;虾去壳、去头,取肌肉部分。经粉碎机粉碎后匀浆,放入-38 ℃的高低温试验箱进行4 h预冷冻,然后用真空冷冻干燥仪对样品进行脱水,冻干后充分研磨,过20目筛。
1.4.2ASE萃取
称取处理后的样品10.00 g,与适量硅藻土放入玻璃研钵中,掺拌均匀并研磨后装入34 mL萃取池(用硅藻土填满池内空隙),进行加速溶剂萃取。萃取溶剂为甲醇,萃取温度为70 ℃,萃取压力为10.34 MPa,静态萃取时间为5 min,循环萃取3次,冲洗体积为60%萃取池体积,氮气吹扫120 s。收集全部萃取液,40 ℃旋转蒸发至近2 mL,用少量甲醇将旋转蒸发瓶底部冲洗2次,合并全部的浓缩液至10 mL的试管里,氮吹至近干,用去离子水定容至1.0 mL,待净化。
1.4.3MSPE净化
将上述样品溶液放入10.0 mL试管中,加入10 mg的GO@nZVI磁性材料,涡旋振荡5 min,利用磁铁将吸附了氟喹诺酮类药物的磁性材料分离,收集在试管底部,弃去上清液。加入0.5 mL纯氨水超声10 min进行洗脱,洗脱溶液在氮气下吹干,然后再溶解于100 μL甲醇中,经0.22 μm滤膜过滤后取10 μL,进行HPLC-MS/MS定量分析。ASE-MSPE萃取流程如图1所示。

图 1 ASE-MSPE萃取流程图Fig. 1 Diagram of accelerated solvent extraction-magnetic solid phase extraction (ASE-MSPE) extraction GO: graphene oxide; nZVI: nanoscale zero valent iron; GO@nZVI: graphene oxide coated nanoscale zero valent iron.
1.5 仪器条件
液相色谱条件:Agilent ZORBAX Eclipse Plus C18色谱柱(100 mm×3.0 mm, 1.8 μm);柱温:35 ℃,流速:0.3 mL/min;流动相A为0.1%(v/v)甲酸水溶液,B为0.1%(v/v)甲酸甲醇溶液。梯度洗脱程序为:0 min (10%B)~1 min (10%B)~7 min (40%B)~10 min (60%B)~11 min (90%B)~12 min (10%B)。
质谱条件:电喷雾离子化(ESI)源,正离子检测模式;多反应监测(MRM)模式进行定量分析。干燥气(N2)温度为300 ℃,流速为5 L/min;雾化气(N2)压力为310 kPa;鞘气(N2)温度为250 ℃,流速为10 L/min;毛细管电压为3 500 V,喷嘴电压为0 V。10种氟喹诺酮的质谱参数见表1, MRM谱图见图2。
2 结果与分析
2.1 ASE萃取条件的优化
萃取溶剂是影响ASE萃取效率的重要因素之一。由于大多数氟喹诺酮类药物为极性化合物,含有氨基和羧基,易溶于水溶液和乙腈,在参考文献[15-18]基础上,比较了2%(v/v)乙酸乙腈、乙腈、pH 7磷酸二氢钾缓冲溶液3种萃取溶剂对10种氟喹诺酮类药物萃取效果。选择乙腈作提取剂时,10种氟喹诺酮类药物的回收率在62.5%~78.1%之间,萃取液较混浊,可能由于样品基质中提取出来的内源有机物较多。磷酸缓冲盐(pH=7)和2%(v/v)乙酸乙腈提取率较好。以磷酸缓冲盐(pH=7)作萃取剂时,10种氟喹诺酮类药物的回收率在80.6%~93.2%之间,但是磷酸缓冲盐提取液呈胶状,这可能由于水产品的蛋白质含量较高,水溶性蛋白质使萃取液呈胶状;以2%(v/v)乙酸乙腈作萃取剂时,10种氟喹诺酮类药物的回收率为76.2%~98.5%,杂质干扰最少,可能由于有机溶剂和水溶液的混合溶剂在一定程度上提高了萃取选择性,减少了杂质的干扰。通过比较不同萃取剂的萃取回收率和提取液的杂质干扰程度,最终选择2%(v/v)酸化乙腈作为ASE萃取剂。

表 1 10种氟喹诺酮类药物的质谱参数

图 2 10种氟喹诺酮的MRM谱图Fig. 2 MRM spectrum of the 10 fluroquinolones
采用正交试验考察了萃取温度(50、70、100 ℃)、萃取时间(2、5、8 min)和循环次数(1、3、5次)3个影响因素在3个水平下对10种氟喹诺酮类药物的提取效率。在空白样品中加入1.0 μg/kg的氟喹诺酮类药物混合标准溶液,根据L9(33)正交试验表进行试验,每个因素设计3个水平,平行3次试验,萃取压力为10.34 MPa,氮气吹扫时间为120 s,冲洗体积为60%。极差(R)分析结果显示,各因素对不同目标物影响的主次顺序为:萃取温度(A)>萃取时间(B)>萃取循环次数(C)。最显著影响因素为萃取温度,根据表2中各因素k值(各因素水平目标物平均回收率的平均值),得到最优组合为A2B2C2,即萃取温度为70 ℃,萃取时间为5 min,循环次数为3次。
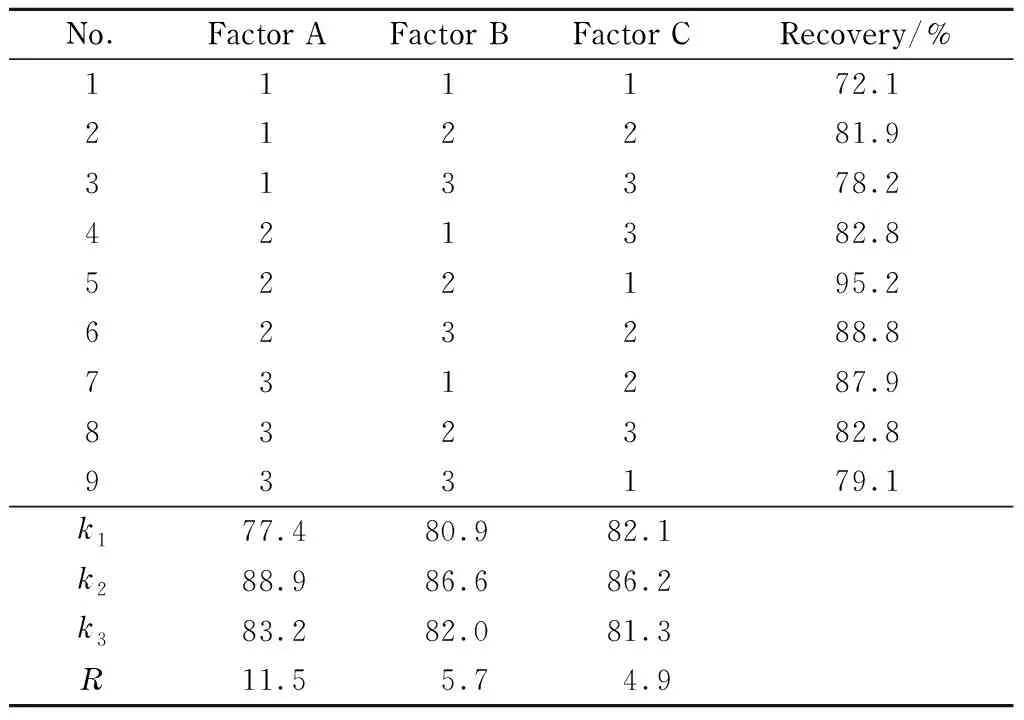
表 2 ASE条件正交试验设计及结果
由于水产品样品基质复杂,为减小基质干扰,样品经ASE萃取后再进一步采用MSPE净化。制备对目标物具有高选择吸附能力的磁性净化材料至关重要,可降低基体干扰,提高分析灵敏度。nZVI具有纳米级粒径、大比表面积、反应活性高、价廉易得等优点[19-21]。由于易被氧化、沉淀,非常细小的nZVI常会发生团聚,导致其有效比表面积明显降低,从而大大降低了吸附活性。为此,可以通过选择合适的碳材料,将nZVI材料负载到不同的碳材料表面,制备nZVI与碳材料的复合物以提高吸附效率。GO含有丰富的含氧官能团,如羟基、羧基、羰基和环氧基等,具有超高比表面积、良好的稳定性、良好的亲水性、分散性和生物相容性、大共轭体系,可以有效吸附与富集氟喹诺酮类抗生素残留[22,23]。本文采用室温自组装法制备GO@nZVI磁性复合材料,相较于其他的磁性材料制备方法,GO@nZVI的制备过程操作简单快速、条件温和,仅需要在室温条件下调节溶液的pH, GO和nZVI可以在静电引力、π-π相互和氢键等作用力下快速结合。同时,制备所得磁性净化复合材料GO@nZVI通过静电引力、π-π相互和氢键等作用力选择性萃取和富集氟喹诺酮类药物,实现进一步净化ASE萃取液的目的,提高检测结果的准确性。
2.2.1磁性净化材料的表征
通过扫描电镜、傅里叶变换红外光谱和X-射线衍射对GO、nZVI和GO@nZVI磁性复合物进行了表征。由图3a可知,GO@nZVI磁性复合物的扫描电镜图中,GO@nZVI粒径约100~500 nm, nZVI被GO片层结构附着后出现团聚,形成蓬松结构,表面粗糙,证明GO@nZVI磁性复合物自组装成功。GO@nZVI的傅里叶变换红外光谱图(见图3b)中含有与nZVI相同的特征峰,而GO的特征峰(-COOH, 1 725 cm-1; C-O-C, 1 224 cm-1; C-OH, 1 042 cm-1)消失,表明GO通过氢键、静电作用力等方式紧密地附着于nZVI的表面,形成GO@nZVI。如图3c所示,GO@nZVI的X-射线衍射谱图中含有与nZVI相同的特征衍射峰,GO中特征衍射峰消失,进一步证明了GO@nZVI是通过氢键、静电作用力等自组装方式复合而成。
2.2.2MSPE条件的优化
使用10 mL的空白加标样品基质ASE萃取液,比较了GO@nZVI用量、吸附时间、洗脱剂、洗脱剂体积和洗脱时间对10种氟喹诺酮类药物的净化效果,平行测定3次。如图4所示,GO@nZVI的最优用量为10 mg,此时FQs的萃取效率达到最大,继续增加用量后基本保持稳定;吸附时间为5 min时,大多数目标物回收率较好;相比较于甲醇、去离子水、氨水溶液(pH 9、11、12),纯氨水的洗脱效果最好。这可能是由于氟喹诺酮类药物为两性分子,易溶于碱性溶液且呈负离子模式,随着pH的增大,与GO@nZVI间静电斥力作用不断增加,氢键作用减弱,使FQs易于从GO@nZVI洗脱下来。进一步考察了洗脱时间(5~15 min)对10种氟喹诺酮类药物回收率的影响。结果表明优化的条件为:GO@nZVI用量为10 mg,吸附时间为5 min,以纯氨水作为洗脱溶剂(0.5 mL),洗脱时间为10 min。
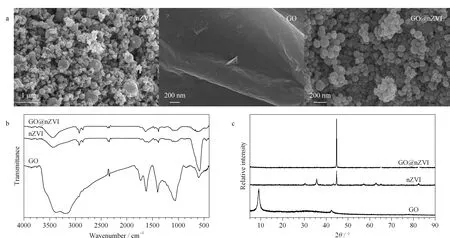
图 3 GO、nZVI和GO@nZVI的(a)SEM图、(b)FT-IR谱图和(c)XRD谱图Fig. 3 (a) Scanning electron microscopy (SEM) images, (b) Fourier transform infrared (FT-IR) spectra and (c) X-rays diffraction (XRD) patterns of GO, nZVI and GO@nZVI

图 4 (a)磁性材料GO@nZVI用量、(b)萃取时间、(c)洗脱剂种类和(d)洗脱时间对MSPE萃取效率的影响(n=3)Fig. 4 Effect of (a) GO@nZVI amount, (b) extraction time, (c) type of desorption solvent and (d) desorption time on magnetic solid-phase extraction (MSPE) efficiency (n=3)
2.3 方法验证
配制10种FQs的混合基质匹配标准溶液,含量分别为1.0、2.0、5.0、10、50、100 μg/kg,在最优条件下进行ASE-MSPE-HPLC-MS/MS分析,得到各目标物的线性方程、线性范围和相关系数。结果表明,在1~100 μg/kg范围内,10种FQs呈现良好的线性关系,相关系数(r2)均大于0.999 5。以3倍和10倍信噪比(S/N)确定检出限(LOD)和定量限(LOQ),分别为0.02~0.29 μg/kg和0.07~0.98 μg/kg(见表3),低于我国规定的最大残留限量要求,满足实际检测需要。
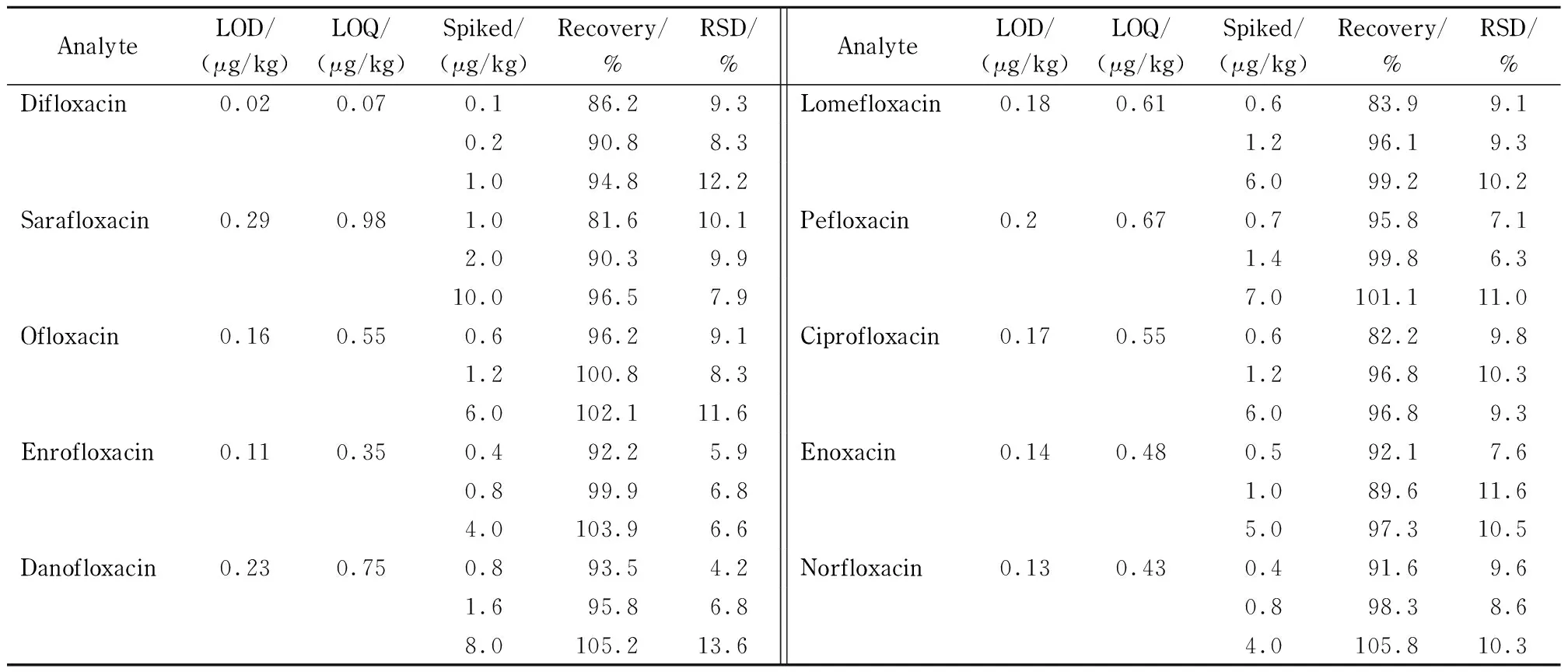
表 3 10种氟喹诺酮抗生素的检出限、定量限、空白加标回收率和相对标准偏差(n=5)
为了考察方法的准确度和精密度,以1倍、2倍、10倍定量限3个添加水平的空白样品进行了加标回收率和RSD的测定,每个水平单独测定6次,回收率的范围在81.6%~105.8%, RSD<13.6%,说明方法具有良好的准确度和精密度。
为了进一步验证本方法的准确度,采用本方法和国标法[24]同时测定同一阳性样品(黄鱼),两种方法分别平行测定5次。以国标法测定的黄鱼中培氟沙星的含量平均值(0.94 μg/kg)作为已知值,本方法培氟沙星的5次测定值分别为1.02、1.14、0.96、0.92和1.01 μg/kg。用t检验法将测定的平均值与已知值进行比较,t测定=1.89,小于t(0.95, n=5)=2.57,说明本方法与国标方法无显著性差异,进一步证明了方法的可靠性和准确性。
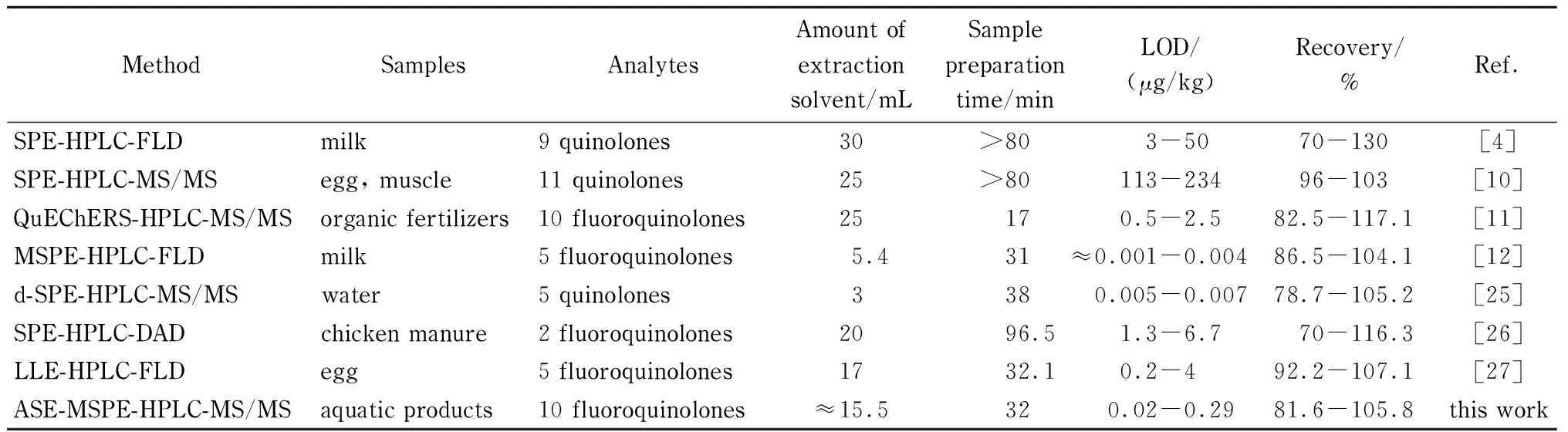
表 4 本方法与文献中氟喹诺酮检测方法的比较
将本方法与文献报道的其他前处理技术结合高效液相色谱检测氟喹诺酮类抗生素药物残留的结果进行比较,其中萃取时间不包括氮吹处理样品的时间,结果见表4。Stoilova等[4]建立了固相萃取-高效液相色谱-荧光法,检测了牛奶样品中9种喹诺酮药物残留,采用液液萃取,通过离心方式进行去蛋白质预处理,再使用HLB固相萃取小柱进行萃取净化,检出限为3~50 μg/kg,该方法的样品前处理时间较长。赵娜等[11]建立了QuEChERS-高效液相色谱-串联质谱法,同时测定有机肥料中10种氟喹诺酮,以酸化乙腈(pH=4.0)为提取剂,经涡旋混合、超声提取,离心分离完成对有机肥料中10种氟喹诺酮的萃取,方法检出限为0.5~2.5 μg/kg,萃取时间为17 min,相对于本方法萃取时间较短,但是单纯QuEChERS方法萃取的净化效果可能较不理想。王成真等[27]建立了液液萃取-高效液相色谱-荧光法,检测鸡蛋中5种氟喹诺酮药物残留,该方法检出限为0.2~4 μg/kg,样品经乙腈沉淀蛋白质,正己烷脱脂,离心分离,上清液后高效液相色谱仪分析。刘艳丽等[12]建立了磁分散固相萃取-高效液相色谱-荧光法,检测牛奶中5种禁用的氟喹诺酮药物残留,方法检出限为1~4 ng/L,采用一步溶剂热法合成了磁性还原氧化石墨烯材料,制备时间>30 min,以磁性材料作为吸附剂,避免了离心过程,萃取时间为16 min,大大缩短了试验时间,但此方法不适合固体样品,且相比于溶剂热法制备,本研究采用室温溶剂制备法,操作更加简单快速。与文献报道方法相比,在回收率相当的情况下,本方法的方法检出限低(0.02~0.29 μg/kg),萃取步骤自动化程度高,且只需在外加磁场作用下即可完成对水产品中10种氟喹诺酮萃取液净化,无需离心和过滤。同样品基质相同或相似的检测方法比较,本方法操作简单快速,萃取溶剂使用较少,所使用磁性萃取材料制备方法简单,使用有机溶剂少,适合水产品中痕量氟喹诺酮残留的检测。
2.5 实际样品测定
选取市售黄鱼、草鱼、黑鱼、明虾和沼虾5种水产品,采用所建立的方法,按照最优实验条件,进行10种氟喹诺酮类药物残留的检测。检测结果显示:黄鱼中培氟沙星的残留量为1.01 μg/kg;黄鱼、草鱼、明虾和沼虾中恩诺沙星的残留量分别为5.16、8.56、1.22和8.07 μg/kg,均低于水产品中氟喹诺酮最大残留限量(10~100 μg/kg)[1];黄鱼和黑鱼中检出禁用氧氟沙星,残留量为1.18、2.59 μg/kg。
3 结论
本文建立了测定水产品中10种FQs残留的加速溶剂萃取-磁固相萃取净化-高效液相色谱-串联质谱分析方法。该方法具有操作简便快速、使用溶剂较少、方法灵敏度高、准确度高、重复性好等特点,可满足我国国家标准中对水产品中FQs限量检测要求,具有实际的应用前景。
