厚朴酚PLGA 纳米粒的制备及其体内药动学研究
2020-11-02张晓千梁丽娜付金芳谈秀凤范明松
张晓千 梁丽娜 付金芳 谈秀凤 范明松
(1.郑州澍青医学高等专科学校, 河南 郑州450064; 2.上海中医药大学, 上海201203; 3.上海雷允上药业有限公司, 上海201401)
厚朴作为临床上广泛使用的中药[1⁃2],其主要活性成分厚朴酚具有中枢神经抑制、中枢性肌肉松弛、抗炎、抗肿瘤、抗氧化、治疗心肌损伤等多种药理作用[3⁃5],但该化合物体内生物利用度较低[6],故促进其体内吸收、提高其生物利用度是急需解决的问题。聚乳酸⁃羟基乙酸共聚物(PLGA)是一种具有良好生物相容性的高分子聚合物,在体内可最终代谢成二氧化碳和水,其纳米粒可提高药物体内稳定性[7],延长药物体内滞留时间,增加药物透膜吸收能力[8],从而改善其生物利用度[7,9⁃10]。因此,本实验制备厚朴酚PLGA 纳米粒,并考察其粒径、Zeta 电位、包封率、载药量、体外溶出、体内药动学,以期为今后相关研究提供依据。
1 材料
QUINTX224⁃1CM 型电子天平[赛多利斯(上海)贸易有限公司];Agilent 1200 型高效液相色谱仪(配置DAD 检测器,美国Agilent 公司);VORTEX⁃2 型涡旋混合器(上海达姆实业有限公司);Nanosep® 超滤离心管(截留分子量10 000 Da,美国Pall 公司);79⁃1 型温控磁力搅拌器(常州金坛良友仪器有限公司);JY92⁃Ⅲ型超声波细胞粉碎机(宁波新知科仪器研究所);RC⁃80 型智能溶出仪(苏州奇乐电子科技有限公司);FD10⁃QX型真空冻干机(上海精密仪器有限公司);Master⁃sizer 型粒度分析仪(英国马尔文仪器有限公司);UGC⁃24C 型氮气吹扫仪(北京优晟联合科技有限公司)。
厚朴酚对照品(批号150520,纯度99.2%,成都普菲德生物技术有限公司);葛根素对照品(批号110752⁃160913,纯度98%,中国食品药品检定研究院)。聚乳酸⁃羟基乙酸共聚物(PLGA,批号20160312,分子量55 000 Da,聚乳酸与聚羟基乙酸比例50 ∶50,济南岱罡生物材料有限公司);0.5%聚乙烯醇(PVA,批号WPE1566D,分子量20 000~30 000 Da,比利时Acros 公司)。透析袋(美国Sigma 公司,截留分子量8 000~14 000 Da)。清洁级SD 大鼠,体质量(300±20)g,购自河南省动物实验中心,动物生产许可证号SCXK(豫)2016⁃0001,在实验室环境下适应1 d 后开始正式研究。
2 方法与结果
2.1 厚朴酚含有量测定
2.1.1 色谱条件 Agilent C18色谱柱(4.6 mm×200 mm,5 μm);流动相乙腈⁃四氢呋喃⁃1.0%冰醋酸(15 ∶1 ∶84);体积流量1.0 mL/min;柱温30 ℃;检测波长320 nm;进样量20 μL。
2.1.2 线性关系考察 称取厚朴酚对照品50 mg,溶于50 mL 乙腈中作为贮备液(1.0 mg/mL),精密量取4.0 mL 至50 mL 量瓶中,流动相定容至刻度,即得对照品溶液(80.0 μg/mL),乙腈依次稀释 至40.0、10.0、1.0、0.2、0.05 μg/mL,在“2.1.1” 项色谱条件下进样测定。以溶液质量浓度为横坐标(X),峰面积为纵坐标(Y)进行回归,得方程为Y=1.623 5X-0.790 8(r=0.999 6),在0.05~80.0 μg/mL 范围内线性关系良好。
2.1.3 方法学考察 取 “2.1.2” 项下0.05、40.0、80.0 μg/mL 对照品溶液,在“2.1.1” 项色谱条件下进样测定,测得厚朴酚峰面积RSD 分别为0.62%、0.25%、0.13%,表明仪器精密度良好。取2.0 mL 厚朴酚PLGA 纳米粒混悬液,置于10 mL量瓶中,4 mL 二氯甲烷超声破乳后乙腈定容至刻度,10 000 r/min 离心30 min,取上清液,于0、2、4、6、8、12 h 在“2.1.1” 项色谱条件下进样测定,测得厚朴酚峰面积RSD 为1.15%,表明溶液在12 h内稳定性良好。平行制备6 份厚朴酚PLGA纳米粒溶液,在“2.1.1” 项色谱条件下进样测定,测得厚朴酚含有量RSD 为1.48%,表明该方法重复性良好。取“2.1.2” 项下贮备液4 mL,乙腈定容至10 mL,即得对照品溶液(400.0 μg/mL),取2.0 mL空白PLGA 纳米粒混悬液至10 mL 量瓶中,平行9 份,加入上 述对照 品溶液0.5、1.0、2.0 mL各3 份,加入4 mL 二氯甲烷超声后乙腈定容至刻度,10 000 r/min 离心30 min,取上清液,在“2.1.1” 项色谱条件下进样测定,测得厚朴酚平均加样回收率分别为98.66%、100.27%、100.02%,RSD 分别1.63%、1.24%、0.96%。
2.2 厚朴酚PLGA 纳米粒及其冻干粉制备
2.2.1 纳米粒 称取厚朴酚10 mg、PLGA 150 mg,溶于15 mL 二氯甲烷中,作为油相;吸取0.5%PVA 溶液40 mL,作为水相,置于磁力搅拌器上,800 r/min 搅拌一段时间,将油相逐滴加到水相中,滴完后超声(150 W)处理5 min(每超声2 s 间隔1 s),得到初乳,减压旋蒸挥去有机溶剂,迅速置于-4 ℃冰箱中固化,即得,并制成混悬液。同法制备空白PLGA 纳米粒混悬液。
2.2.2 冻干粉 将厚朴酚PLGA 纳米粒混悬液分成若干份,12 000 r/min 离心20 min,弃去上清液,收集沉淀,蒸馏水定容至40 mL,加入3%甘露醇作为冻干保护剂,-60 ℃下预冻36 h 后抽真空,冷冻干燥48 h,即得。
2.3 载药量、包封率测定 采用超滤离心法。取2.0 mL 厚朴酚PLGA 纳米粒混悬液至10 mL 量瓶中,4 mL 二氯甲烷超声破乳后,乙腈定容至刻度,8 000 r/min 离心30 min,取上清液,在“2.1.1”项色谱条件下进样测定,计算厚朴酚总含有量m总;精密量取2.0 mL 纳米粒混悬液至超滤离心管中(截留分子量30 kDa),12 000 r/min 离心20 min,在“2.1.1” 项色谱条件下进样测定,计算游离厚朴酚含有量m游离。再计算载药量、包封率,公式分别为载药量= [(m总-m游离)/(m总+m纳米粒)]×100%、包封率= [(m总-m游离)/m总]×100%,其中m纳米粒为纳米粒总质量。
按“2.2” 项下方法平行制备3 批厚朴酚PLGA 纳米粒及其冻干粉,测得前者平均包封率为(75.36±0.38)%,载药量为(4.61±0.27)%;后者包封率下降至(71.02±0.41)%,载药量下降至(4.42±0.32)%,质量分数为0.51%(以厚朴酚计)。
2.4 形态观察 取适量厚朴酚PLGA 纳米粒混悬液,2%磷钨酸溶液染色后置于透射电镜(TEM)下观察其基本形态,结果见图1。由此可知,纳米粒子之间无粘连,基本呈球形或类球形。

图1 纳米粒TEM 图Fig.1 TEM image for nanoparticles
2.5 粒径、Zeta 电位测定 取3 份厚朴酚PLGA纳米粒混悬液,每份100 μL,超纯水稀释40 倍后测定粒径、Zeta 电位,超纯水复溶其冻干粉后也同法测定,结果见表1。由此可知,与纳米粒比较,其冻干粉粒径升高,Zeta 电位绝对值降低。

表1 冻干前后纳米粒平均粒径、Zeta 电位测定结果(n=3)Tab.1 Results of average particle size and Zeta potential determination of nanoparticles before and after lyophilization(n=3)
2.6 体外释药研究 采用透析袋法。取厚朴酚PLGA 纳米粒冻干粉和原料药(含5 mg厚朴酚),加入3 mL 0.5%SDS 溶液制备混悬液,转移至透析袋中(截留分子量8 000~14 000 Da),两端尼龙绳扎紧,设定溶出仪转速、温度分别为100 r/min、(37±1)℃,以900 mL 0.5%SDS 溶液为释放介质,于0.25、0.5、1、1.5、2、2.5、3、4、6、8、12、24 h 各自动取样2 mL,并补加2 mL 蒸馏水以保持总体积不变,溶液离心(4 000 r/min)20 min 后,在“2.1.1” 项色谱条件下进样测定,结果见图2,可知纳米粒在前4 h 释药较快,之后趋于平稳。再分别采用零级、一级、Weibull、Higuchi 方程进行拟合,发现Weibull 方程拟合效果最好(R2=0.978 3)。
2.7 药动学研究
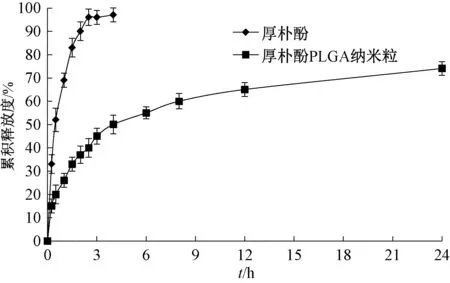
图2 样品体外释药曲线Fig.2 In vitro drug release curves for samples
2.7.1 给药及血样采集 取大鼠12 只,随机分为2 组,给药前禁食12 h,自由饮水,分别灌胃给予厚朴酚及其PLGA 纳米粒混悬液(5 mg/mL),给药剂量50 mg/kg(以厚朴酚计)。乙醚麻醉大鼠后,用肝素浸润的玻璃毛细管于0.25、0.5、1、2、2.5、3、4、6、10、12 h 眼眶后静脉丛采血各0.2~0.3 mL,置于肝素浸润的离心管中,立即用棉球覆盖在取血眼眶处止血。血液在2 500 r/min下离心2 min 后,取上层血浆,低温保存。
2.7.2 血浆样品处理 精密称取葛根素对照品10 mg,溶 于 100 mL 乙腈中,乙腈稀释至200 ng/mL,即得内标溶液。精密吸取 “2.7.1”项下解冻后血浆200 μL、内标溶液100 μL 于离心管中,加入1.0 mL 乙酸乙酯涡旋振荡3 min,静置1 min 后,继续涡旋振荡3 min,3 000 r/min 离心3 min后,取上层有机相,45 ℃下氮气吹干,加入200 μL 乙腈,8 000 r/min 离心5 min,取20 μL上清液,在“2.1.1” 项色谱条件下进样测定。
2.7.3 线性关系考察 精密吸取“2.1.2” 项下1.0 μg/mL 对照品溶液适量,乙腈依次稀释至800、500、100、50、20 ng/mL,各精密量取200 μL,与100 μL 内标溶液置于同一离心管中,45 ℃氮气吹干,加入200 μL 空白血浆涡旋5 min,取20 μL,在“2.1.1” 项色谱条件下进样测定。以溶液质量浓度为横坐标(X),厚朴酚、内标(葛根素)峰面积比值为纵坐标(Y)进行回归,得方程为Y=0.079 12X+0.008 4(r=0.992 2),在20~1 000 ng/mL 范围内线性关系良好。
2.7.4 方法学考察 取空白血浆、空白血浆+对照品+内标(按“2.7.3” 项下方法制备)、处理后血浆样品(按“2.7.2” 项下方法制备)溶液适量,在“2.1.1” 项色谱条件下进样测定,结果见图3。由此可知,厚朴酚与内标分离度较高,血浆内源性物质不干扰测定,表明该方法专属性良好。
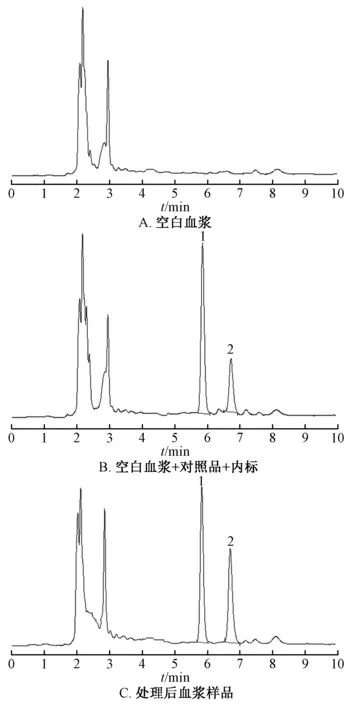
图3 厚朴酚HPLC 色谱图Fig.3 HPLC chromatograms of magnolol
取处理后血浆样品溶液适量,室温下于0、2、4、6、12、24、36 h 在“2.1.1” 项色谱条件下进样测定,测得厚朴酚含有量RSD 为9.67%,表明溶液在36 h 内稳定性良好。取“2.7.3” 项下20、500、1 000 ng/mL 空白血浆+对照品+内标溶液,在“2.1.1” 项色谱条件下进样测定6 次,测得厚朴酚含有量RSD 分别为1.83%、0.82%、0.71%,表明仪器精密度良好。取 “2.7.3” 项下800、500、100 ng/mL 对照品 溶液各200 μL,加 入100 μL内标溶液,氮气吹干后加入200 μL 空白血浆重新溶解,按 “2.7.2” 项下方法处理,在“2.1.1” 项色谱条件下进样测定3 次,测得厚朴酚平均加样回收率分别为95.18%、90.83%、91.66%,RSD 分别为8.43%、5.69%、7.10%。取20 ng/mL 对照品溶液,乙腈逐步稀释,测得定量限(S/N=10)为12 ng/mL,检测限(S/N =3)为5 ng/mL。
2.7.5 结果 图4、表2 显示,纳米粒tmax、Cmax、AUC0~t、AUC0~∞高于原料药(P<0.05,P<0.01),并且相对生物利用提高至2.17 倍。

图4 样品血药浓度⁃时间曲线Fig.4 Plasma concentration⁃time curves for samples
表2 样品主要药动学参数(, n=6)Tab.2 Main pharmacokinetic parameters for samples(, n=6)

表2 样品主要药动学参数(, n=6)Tab.2 Main pharmacokinetic parameters for samples(, n=6)
注:与厚朴酚比较,∗P<0.05,∗∗P<0.01。
3 讨论
在药动学研究中,血浆样品的处理方法对实验结果准确性有较大影响。课题组前期曾先用甲醇、乙腈等沉淀蛋白,再用乙酸乙酯萃取,但发现样品回收率仅为54.11%~68.79%;用乙酸乙酯直接萃取时,回收率反而升至90.69%~95.42%。研究表明,厚朴酚与血浆蛋白之间可能会产生静电作用力、氢键作用力等,导致有机溶剂沉淀蛋白时厚朴酚与血浆蛋白结合较紧密[11]。因此,最终确定处理方法为乙酸乙酯直接萃取,而且操作过程大大简化。
厚朴酚生物利用度低,可能与其水溶性差、透膜吸收能力有限、稳定性不理想等因素有关[12⁃15],将其制成PLGA 纳米粒后,tmax显著延长,Cmax显著升高,相对生物利用度提高至2.17 倍。其中,tmax延长的原因一方面与纳米粒易吸附在胃肠道黏膜而增加滞留时间有关,另一方面也与PLGA 聚合物纳米粒子缓释作用相关;Cmax、相对生物利用度升高的原因是由于纳米粒比表面积明显变大,增加了药物与胃肠道的接触面积,而且它可在胃肠道中与具有淋巴结构的Peyer’ s parches 聚集,通过M 细胞进入血液循环,从而促进药物高效吸收[16⁃20]。但厚朴酚PLGA 纳米粒口服生物利用度的提高程度还受到酶解、制剂稳定性、透膜吸收能力等因素的影响[10],可能会导致体内外相关性不一致。另外,PLGA 纳米粒注射后还会受到网状内皮细胞清除、单核巨噬细胞吞噬等因素的作用,其绝对生物利用度的提高程度仍有待进一步研究[21]。
