基于生物信息学分析筛选肾上腺皮质癌核心基因
2020-10-26张德莲蔡昕添李南方
张德莲 蔡昕添 洪 静 朱 晴 吴 婷 李南方
ScreeningHubGenesofAdrenocorticalCarcinomaBasedonBioinformaticsAnalysis.ZhangDelian,CaiXintian,HongJing,etal.HypertensionCenterofthePeople′sHospitalofXinjiangUygurAutonomousRegion,HypertensionInstituteofXinjiang,NHCKeyLaboratoryofHypertensionClinicalResearch,Xinjiang830001,China
AbstractObjectiveTo study hub genes of adrenocortical carcinoma, and to explore possible prognostic evaluation and screening of molecular biomarkers for diagnosis by bioinformatics methods, which provides bioinformatic evidence support for potential therapeutic targets of adrenocortical carcinoma.MethodsThe data of GSE14922, GSE19750 and GSE90713 chips were downloaded from GEO database, and a series of bioinformatics methods were used to process, analyze and screen the differential genes of GSE14922 chips. Then select hub genes through CytoHuhha plug-in, and finally use the online analysis tools GEPIA and GSE19750, GSE90713 chip data to carry out survival analysis and diagnostic test analysis of hub genes.ResultsTotally 90 differential genes were obtained from the GSE14922 chip. The 6 hub genes obtained through the CytoHuhha plug-in are: BUB1, CDK1, CENPF, NDC80, ASPM and DLGAP5. The results ofKaplan-Meiersurvival curve analysis showed that the above hub genes have a significant impact on the diagnosis and survival prognosis of adrenocortical carcinoma patients. The results of ROC analysis show that the above hub genes have better diagnostic significance for patients with adrenocortical carcinoma.ConclusionThe 6 hub genes obtained through bioinformatics analysis play an important role in the progression of adrenal cortical cancer, and provide a new theoretical basis for revealing the potential diagnosis, prognostic markers and therapeutic targets of adrenal cortical cancer.
KeywordsAdrenocortical carcinoma; Hub genes; Biomarkers; Bioinformatics
肾上腺皮质癌(adrenocortical carcinoma,ACC)是一种罕见的恶性内分泌系统肿瘤,年发生率为(0.7~2.0)/百万。ACC的预后极差,即使早期诊断并通过手术将其切除的情况下,其复发和转移的风险仍然很高,5年总生存率仍低于40%[1]。近年来尽管ACC的临床诊治流程取得了巨大的优化,但由于早期诊断困难及缺乏有效的治疗策略,只有少数患者可获得显著的生存益处[2]。越来越多的证据表明,多基因的异常表达参与了ACC的癌变与进展[1]。因此,探究ACC的发生、增殖和复发的分子机制,发现新的治疗靶点,从而制定出有效的诊断和治疗策略,对于改善ACC患者的预后具有极为重要的作用。近年来微阵列技术和生物信息学分析被广泛应用于检测与分析基因组学的综合表达情况。本研究从GEO数据库下载了3份基因芯片数据集,并对其进行分析,以获得ACC癌组织与非癌组织之间的差异基因(differentially expressed genes, DEGs)。随后,通过生物信息学分析进一步探讨ACC发生的分子机制,并揭示ACC潜在的诊断、预后标志物和治疗靶点。
材料与方法
1.原始芯片数据来源:本研究中原始DNA微阵列数据是从GEO数据库中获得的。根据不同平台中的注释信息,将探针信号转化为的相应基因名字。共获得3份芯片数据集(GSE14922、GSE19750和GSE90713)。其中,基于GPL6480平台(Agilent-014850 Whole Human Genome Microarray 4x44K G4112F)的GSE14922芯片,包含4个正常的肾上腺皮质组织、8个腺瘤组织和4个原发性肾上腺皮质癌组织;基于GPL570平台[(HG-U133_Plus_2) Affymetrix Human Genome U133 Plus 2.0 Array]的GSE19750芯片,包含4个正常的肾上腺皮质组织和44个原发性肾上腺皮质癌组织;基于GPL15207平台(Affymetrix Human Gene Expression Array)的GSE90713芯片,包含5个正常的肾上腺皮质组织和58个原发性肾上腺皮质癌组织。
2.芯片数据预处理和差异基因筛选:利用R语言软件(3.6.3版)中的Affy包对GSE14922芯片的原始数据进行初步分析,并将微阵列文件转换成基因表达谱数据。然后使用稳健的多阵列平均法来标准化表达谱数据。此外,使用Limma软件包计算和分析样本组与对照组之间的差异基因,采用t检验和fold-change(FC)对癌组织和正常组织间的基因表达进行显著性分析。然后,使用Benjamini-Hochberg法计算调整后的P值(FDR)。GSE14922芯片DEGs筛选的最终结果以火山图的形式展现。统计学差异标准为FDR<0.01,|logFC|≥2。
3.蛋白质相互作用网络的构建与核心基因的筛选:蛋白质相互作用网络分析是一种识别各种蛋白质之间联系关系的方法。为了进一步了解AAC分子机制,将DEGs列表输入到STRING(版本11.0)数据库的搜索工具中。利用该工具,选择相互作用关系评分>0.7的蛋白质构建相互作用网络[3]。使用Cytoscape 3.7.2软件对从STRING数据库中获得的蛋白质相互作用网络进行可视化处理,并利用CytoHuhha插件的Stress算法、MCC算法、DMNC算法和MNC算法从蛋白质相互作用网络中计算排名前10的基因[4]。最后利用韦恩图取交集,将4种算法皆排名前10的基因选作核心基因。
4.核心基因的生存预后分析:GEPIA在线分析工具是一个基于TCGA数据库和GTEx项目的在线分析网站。通过使用GEPIA在线分析工具对所筛选到的核心基因进行生存分析,以检查上述核心基因表达水平的改变与患者的存活率之间的关系,绘制核心基因的Kaplan-Meier生存曲线并计算高低表达间的生存是否存在统计学差异,以P<0.05为差异有统计学意义[4]。
5.核心基因的诊断价值评价:本研究利用GSE63060和GSE63061数据集的数据对上述核心基因的诊断价值进行了评价。本研究采用R语言中的pROC软件包绘制ROC曲线并通过计算ROC曲线下面积(AUC)来评价每一个核心基因的诊断性能。当AUC值>0.7时,则该核心基因被认为具有较好的诊断性能。
结 果
1.芯片数据预处理和差异基因筛选的结果:从GSE14922数据集中共获得到16个样本的芯片检测数据。在进行质量控制及数据的预处理后,对每个芯片的序列分别进行了分析,共获得了19553个基因的表达水平信息。根据FDR<0.01和|logFC|≥2的标准,共选择了90个DEGs,包括53个上调基因和37个下调基因,火山图见图1。

图1 差异表达基因的火山图红色.显著上调基因;绿色.显著下调基因;黑色.表达差异无统计学意义基因
2.蛋白质相互作用网络的构建与核心基因的筛选:利用STRING数据库对90个差异表达基因进行了蛋白质相互作用的网络分析并将分析结果导入Cytoscape软件中,得到的相互作用网络由87个节点以及453条边组成,详见图2A。利用Cytoscape软件的CytoHuhha插件分析筛选核心基因,在4种算法的计算下均排名前10的DEGs被认为核心基因。最终通过韦恩图取交集获得肾上腺皮质癌的核心基因有BUB1、CDK1、CENPF、NDC80、ASPM和DLGAP5,详见图2B。
3.核心基因的生存预后分析:利用在线分析工具GEPIA对上述核心基因进行生存预后分析,详见图3。从Kaplan-Meier生存曲线的结果中可以认为BUB1基因(HR=7.7,P=0.000)、CDK1基因(HR=11,P=0.000)、CENPF基因(HR=4.6,P=0.000)、NDC80基因(HR=4.9,P=0.000)、ASPM基因(HR=12,P=0.000)和DLGAP5基因(HR=7.8,P=0.000)对肾上腺皮质癌患者的生存预后具有显著影响。
4.核心基因的诊断价值评价:在GSE63060和GSE63061数据集的数据中BUB1基因(AUC=0.963)、CDK1基因(AUC=0.977)、CENPF基因(AUC=0.989)、NDC80基因(AUC=0.955)、ASPM基因(AUC=0.957)和DLGAP5基因(AUC=0.966)对在ACC患者与对照人群中表达水平的差异具有较好的诊断意义,可用于ACC患者的诊断,详见图4。
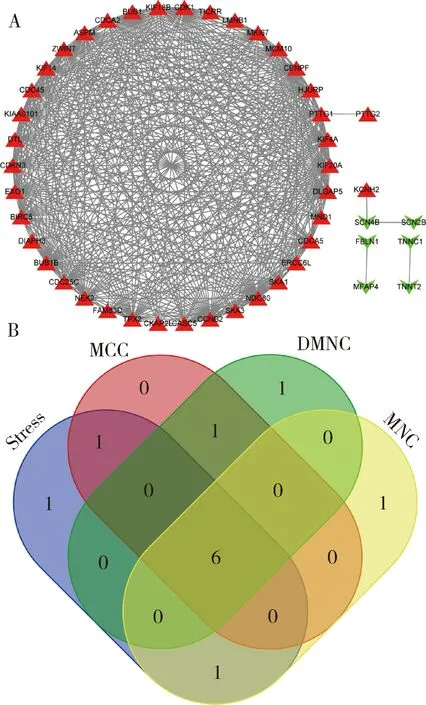
图2 蛋白质相互作用网络图与核心基因的筛选A.蛋白质相互作用网络图(红色模块代表表达上调蛋白质,绿色模块代表表达下调蛋白质);B.韦恩图

图3 肾上腺皮质癌相关核心基因生存预后分析A.ASPM基因;B.BUB1基因;C.CDK1基因;D.CENPF基因;E.DLGAP5基因;F.NDC80基因

图4 6个核心基因诊断ACC的ROC曲线
讨 论
ACC是一种罕见且侵袭性极高的癌症[1]。据报道儿童期AAC的发病绝大多数与TP53的种系突变密切相关,但对于成人ACC的发病机制和主要分子机制却了解甚少[5]。越来越多的数据表明,成人ACC的发病是多基因参与、多通路相互作用的结果[6]。因此,需要更好地理解ACC发病机制和相关分子机制,以揭示ACC潜在的诊断、预后标志物和探索新的治疗靶点,从而更积极地改善患者预后。
BUB1基因及其重要旁系同源物BUB1B基因均参与编码丝氨酸/苏氨酸蛋白激酶,在有丝分裂中发挥核心作用[7]。编码的蛋白质部分通过将有丝分裂检查点复合物的成员磷酸化并激活纺锤体检查点来发挥功能[8]。BUB1基因还在DNA损伤修复、抑制后期促进复合物/环体的活化中发挥重要作用[9,10]。目前的研究表明,BUB1基因的突变与几种非整倍性相关的肿瘤密切相关[9]。CDK1基因主要编码细胞周期蛋白依赖性激酶1,是一种高度保守的蛋白激酶复合物的催化亚基[10]。因其对于真核细胞周期的G1/S和G2/M相变至关重要,所以又被称为M期促进因子。其主要参与细胞周期调控、DNA损伤修复和检查点转录等生物学过程[11]。目前已有的研究表明与CDK1相关的疾病主要包括视网膜母细胞瘤和乳腺癌,其主要参与途径包括卵母细胞减数分裂和ERK信号转导[12]。位于染色体1q41上的CENPF基因所编码蛋白质主要起着丝粒-线粒体复合体的作用,并且可作为相间G2期间核基质的重要组成部分。作为染色体分离的调节剂,CENPF以细胞周期依赖性方式表达。有研究表明CENPF通过对信号转导、葡萄糖代谢和表观遗传调控等作用,在腺体细胞性肿瘤进程中扮演着重要角色[13]。
NDC80基因是一种蛋白质编码基因,其编码的蛋白质由N末端微管结合结构域和与复合物其他成分相互作用的C末端卷曲螺旋结构域组成[14]。NDC80通过介导纺锤体装配检查点信号和染色体比对在染色体分离中发挥重要作用。染色体分离功能障碍是染色体不稳定的重要原因之一,而染色体的不稳定性是肿瘤细胞的共同特征之一,是肿瘤形成与进展的重要机制[15]。ASPM基因位于1q31染色体上,编码异常纺锤体微管装配体蛋白。ASPM基因的表达对于胚胎成神经细胞中正常的有丝分裂纺锤体功能和神经发生的调节至关重要[16]。
有研究表明,ASPM与肝细胞癌、卵巢癌、胰腺癌和前列腺癌的不良预后显著相关[17]。DLGAP5基因主要编码有丝分裂纺锤体蛋白,该蛋白可促进微管蛋白聚合物的形成,从而在微管的末端附近形成微管蛋白片[18]。DLGAP5作为细胞周期调节剂参与多种癌症的形成和发展,目前已有报道DLGAP5在结肠癌、乳腺癌、肝癌和膀胱癌等癌症中存在过度表达的情况,并且该基因及其产物可能是潜在的肿瘤治疗靶点[19]。
综上所述,本研究通过基因组学高通量测序结合生物信息学分析的方法对4个正常的肾上腺皮质组织、8个腺瘤组织和4个原发性肾上腺皮质癌组织进行了全基因组基因的表达分析,以探索与ACC发生和进展相关的差异表达基因,共发现90个DEGs。通过构建PPI网络并通过4种不同算法获得6个核心基因。采用不同数据库和数据芯片进行预后分析与诊断试验分析进一步证实,BUB1、CDK1、CENPF、NDC80、ASPM和DLGAP5基因的表达水平既可用于评估ACC患者的预后情况又具有良好的预测诊断性能。本研究为探讨ACC的分子细胞机制以及潜在治疗靶点提供了生物信息学证据支持,并意味着其可作为提高ACC诊断准确性及靶向药物开发的潜在生物学标志物。虽然本研究具有上述优点,但仍存在部分局限性。首先,缺乏核心基因与临床病理数据的相关性分析。其次,本研究未进行Western blot法检测分析或免疫组化染色来评价核心基因编码蛋白在ACC肿瘤组织中的表达情况。因此,在未来的研究中需进一步收集临床数据与病理学证据并进行Western blot法检测分析及免疫组化染色,进一步验证生物信息学方法分析结果的准确性。
