Defect levels in d-electron containing systems: Comparative study of CdTe using LDA and LDA + U
2020-10-22YuanYinYuWangGuangdeChenandYelongWu
Yuan Yin , Yu Wang Guangde Chen and Yelong Wu
1College of Physics and Optoelectronic Technology, Advanced Titanium Alloys and Functional Coatings Cooperative Innovation Center,Baoji University of Arts and Sciences, Baoji 721013, China
2Department of Applied Physics and the MOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter,Xi’an Jiaotong University, Xi’an 710049, China
Abstract: The defect properties in d-electron containing materials will be strongly influenced by the non-negligible on-site Coulomb interactions. However, this has been omitted in the current widely adopted standard first-principles calculations, such as LDA, leading to a large deviation of calculated results. Therefore, as a comparative case study, in this paper the defects of CdTe are investigated by first-principles calculations including standard LDA and LDA + U, and we find that LDA + U gives more accurate formation energies of the neutral point defects than the standard LDA. The same trend can be found in transition energies of the charged state defects as well. These comparative analyses show that LDA + U gives better results for the defects of CdTe than the standard LDA and requires less computing resource than LAPW, indicating it should have huge potential to model supercells with large number of atoms and strong electron interactions. Moreover, a new anion interstitial defect structure is found to be more stable than the well-known tetrahedron central anion interstitial defect structure
Key words: defects; LDA; LDA + U; formation energy; transition energy; first-principles
1. Introduction
Many semiconductors such as CdTe, ZnO, AlN and their related II–VI and III–V compounds would not be very useful without being doped[1−13]. The performance of these materials depends critically on their electrical properties, which are dominated by the formation of defects, including both point and extended defects. It has been revealed that cadmium telluride (CdTe) is the only II–VI compound that can be doped relatively easily, either p-type or n-type[14,15]. Its alloys are adopted to fabricate optoelectronic applications such as solar cell[1,2]and x- and γ-ray semiconductor detectors[3−5]. To implement these applications, it is necessary to understand the formation of defects in CdTe. As a powerful method, density-functional theory (DFT) is used to calculate the structural and electrical properties of semiconductors. However, different calculation methods have various results on defects in CdTe.
In consideration of the full-electronic potential, linearized augmented plane wave (LAPW)[16,17]is a relatively accurate method to calculate defects in first-principle calculation.In addition, local density approximation (LDA)[18,19]can also be used to solve these issues with relative less computational demanding comparing with LAPW. The calculated band gaps of CdTe with different calculation methods vary, 1.48 eV[20]by LAPW at the theoretical lattice constant, which is in good agreement with the experimental value of 1.61 eV[21],while only 0.63 eV by standard LDA. This is largely because standard LDA overestimates the delocalization of Cd-4d electrons, which pushes the valance electrons (the Te-5p valence band) up. For general systems without d or f electron, the electron interaction is not so strong that standard LDA method can meet the requirement to handle their exchange-correlation potentials.
For systems with strong electron interaction, the calculated values are not accurate and we usually adopt LDA +U[22]approach with Hubbard-like model[23]to correct the calculated results. There are two parameters that are very important for the approach of LDA + U to describe non-integer or double occupations of states, these are (1) U, which is used to describe the strength of the on-site Coulomb interaction,and (2) J, which is used to adjust the electron exchange interaction. To be simple, Dudarev’s method[24]can be used, in which these two parameters can be combined into a single efficient one, Ueff= U – J, and this effective parameter Ueffis typically referred to as U. For CdTe, from the previous work about determining U value[25,26], we find that the lattice constant becomes closer to the experimental value with the U value increasing to 13 eV expressly, while the U value decreases to 7 eV, the result of LDA + U calculation matches well with experimental photoemission spectra. In this work,therefore, the density of states calculations was operated at U = 7 eV.
In this paper, we systematically calculate the lattice constants, model structures, electronic band structures, formation energies and transition energies of defects in CdTe by different calculation methods. The target is to find out a method which can reflect the physical properties of defects better in CdTe. For most of the cases, LDA + U gives better results than the standard LDA. Since this demands a low computing power relative to LAPW, the LDA + U approach is an attractive means to study the supercells with large number of atoms and strong electron interactions.
2. Method of calculation
In this paper, DFT was used to perform all the structural optimizations, energy band structures and total energy calculations. The exchange-correlation functional was handled by both standard LDA and LDA +Umethods and the VASP code was used in all cases[27−29]. During the whole calculations, all the atoms were allowed to relax fully until the force acting on them became less than 0.03 eV∕Å. In consideration of the size and boundary effects, a large enough model was often adopted to simulate defect structure, in which we performed CdTe of zinc-blende structure with 64 atoms, and the defect structures were modeled by putting into or taking out, or substituting one atom at the center of the periodic supercells. During the calculations, the energy cutoff was set at 350 eV, and 3 × 3 × 3k-point grid was used.
It is well-known that formation energies of the defects can reflect the degree of difficulty or ease commendably during the defect formation in bulk materials. The larger the formation energy is, the more difficultly the defect forms, and vice versa. To determine the defect formation energy and defect transition energy levels, first, it is necessary to identify the defects with low formation energies for every position of the Fermi level and every possible stoichiometry. Second, we need to calculate the total energyE(α,q) of the supercell containing the relaxed defectαin charge stateq. In addition, we also need to calculate the total energyE(host) for the same supercell in the absence of the defect, as well as the total energyEiof the basic composition element. The defect formation energy depends not only on the atomic chemical potentialμibut also on the electron Fermi energyEf. From these parameters, the defect formation energy ΔH(α,q) can be defined as follows

whereniis the number of elements which constitute the supercell, which added to (ni> 0) or taken from (ni< 0) the bulk crystal to create the defect, andqis the number of electrons which transfer from the supercell to the reservoirs while forms the defect cell;μiis the atomic chemical potential with constituentireferenced to elemental solid or gas withEi;Evis the valance-band maximum (VBM);μeis the electron chemical potential which was measured relative to the VBMEv. The last term (μe+Ev) represents the electron Fermi energyEf.
In addition, the transition energyεα(q/q′) for the defectαwhich transfers fromqcharge state toq′charge state can be obtained by

The calculation results are sensitive to thek-points sampling, thusΓ-point-only approach[30]is often used to avoid this problem. To combine the advantages of specialkpoints andΓ-point-only approaches, we adopt a new hybrid scheme[31]to calculate the transition energy. In this new scheme, for acceptor defect (q< 0), the transition energy with respect to VBM is given by:

For donor defect (q> 0), the ionization energy referenced to CBM is given by:

Under equilibrium growth conditions, there are some thermodynamic limits on the achievable value of the atomic chemical potentialsμiin Eq. (1). By means of some certain conditions, we can obtain the formation energy simply without using the atomic chemical potentialsμi, then after deducing from Eqs. (1) and (2), the formation energy of charge state is given by:
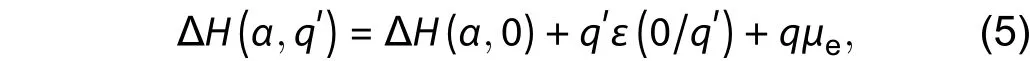
where ΔH(α,) is the defect formation energy of neutral defects, this equation gives a perfect linear relation that the formation energy of charge state ΔH(α,q′) changes with the value of electron chemical potentialμe, which has been used successfully to research defects in a variety of semiconductors[6,7,9].
3. Results and discussion
There is a slight discrepancy on the lattice constant for undoped CdTe by different calculation methods, and the result is given as follows: it is 6.421 Å by standard LDA, and 6.408 Å by LDA +U, and 6.541 Å by LAPW[9].
It is widely-believed that band gap is always considered as the most important aspect to describe the electronic properties of semiconductors for optoelectronic applications. LAPW predicts a band gap of 1.48 eV (1.59 eV at the experimental lattice constant), which agrees well with the experimental value[21]of 1.61 eV. Standard LDA gives a band gap of 0.63 eV for CdTe, which is significantly lower than the experimental value. However, LDA +Uproduces a band gap of 0.96 eV[32].The Cd-4d levels shift down from the VBM, which induces the p–d coupling to become weak, thus the valence band and conduction band both drop down. As the valence band drops down more seriously than the conduction band, we can observe that the band gap increases comparing with the band gap of standard LDA.
3.1. Formation energies of the neutral point defects
The calculated defect formation energy ΔH(α,q) at neutral charge state (q= ) andμi= 0 by different approaches(LAPW[9], LDA, and LDA +U) are shown in Table 1. In addition, one can deduce the defect formation energy ΔH(α,q)with different chemical potentials through Eq. (1).
In this paper, the results of defect formation energy calculated by LAPW method are obtained from Wei’s work[9]. Theresults of standard LDA and LDA +Uapproaches are from our calculations. From Table 1, we find that the majority formation energies of LDA +Uare closer to the values of LAPW than that of standard LDA.

Table 1. Calculated defect formation energy Δ H(α,q) [through Eq.(1)] of point defects for tetrahedron structure at neutral charge state(q= ) and μi = 0, the unit of defect formation energies is eV.
For interstitial defects, two cases always should be considered in the zinc-blende structure, these areandwhich indicate the interstitial A atom is surrounded by anion atoms or cation atoms[33]. In this work, cation interstitial defects includeandFor bivalent Cd and Cu interstitials, we find that the results are almost the same with each other by different approaches. While for univalent Na interstitial, both standard LDA and LDA +Uresults have obviously lower formation energies than LAPW. As shown in Table 1, the results of formation energies ofdefect are 3.52, 2.07 and 1.94 eV with these different methods, respectively. In LAPW calculations[9]forthe stable structure has the Te atom at the center of the tetrahedron, as shown in Fig. 1(a). However, when we operate the simulation process with standard LDA and LDA +Uapproaches, we find a much more stable structure, in which the anion atom Te and two cation atom Cds are almost in a line, see Fig. 1(b),while the origin of bonding has been explained[34].
For anti-site defects, it is obvious that the result of LDA +Uis inferior to that of standard LDA for group-IB elements substituting the Cd site,(IB = Cu, Ag, Au withdelectrons and without considering plusU). However, LDA +Uis distinctly superior to standard LDA when the group-IIIA elements substitute Te site(IIIA represents Br, In withdelectrons without considering plusU). For Te vacancy and Cd vacancy, the results of LDA +Uare closer to LAPW results than standard LDA as well. In general, we can make a conclusion that the majority of calculated formation energies by standard LDA and LDA +Uare lower than LAPW.
3.2. Transition energies of the charged state defects
In this section, we discuss and analyze the calculated optical transition energies of charged state defects in CdTe. Figs. 2 and 3 describe the transition energy positions of CdTe, which are calculated by different methods. Similarly, Table 2 presents the calculated acceptor defect (q< 0) transition energies and Table 3 gives the donor defect (q> 0) transition energies of tetrahedron CdTe. These results are acquired by using Eqs. (3) and (4). One can also deduce the formation energies of charged state defects from Eqs. (1) and (2). Meanwhile,k-point, core level, and electrostatic correction were also taken into consideration during the transition energies calculation.
Table 2 shows that the intrinsic defect Teia(0/–2) has a deep acceptor level above the VBM[25,26]. The transition energies calculated by standard LDA and LDA +Uare 1.795 eV and 1.993 eV, which are higher than the result of LAPW(0.57 eV) overly. As mentioned earlier, the obvious discrepancy can be accounted for the different Teiastable structures.In our structure, the defect level goes up to conduction band and even over it; that is to say, the transition energy is greater than band gap. For the previous structure, Te is at the center of the tetrahedral, and the defect level is in the band gap.
For the intrinsic defect VCd, which has relative shallow transition energy levels with respect toIt is the most common intrinsic acceptor in CdTe, whose (0/–1) transition energy level is high with 0.13 and 0.173 eV by LAPW and LDA +U, while standard LDA gives a value of 0.091 eV which is small enough to reach high hole density at room temperature. After comparing with the experimental results, LDA +Umight be, in this case, the worst method.
For extrinsic acceptor, the results of(IB represents Cu, Ag, Au, Na) and(IIIA represents Sb, As, P, N) by standard LDA and LDA +Uindicate these defects all have relative shallow (0/–1) levels compared withand there is no apparent difference between these methods. As shown in Table 2,a distinct phenomenon is that most of the calculated (0/–1)transition energy results obtained by LDA +Uare improved obviously relative to that of standard LDA, but some become worse slightly, such as AuCd, NaCdand BiTe, whose results of standard LDA are closer to that of LAPW. No matter how disparate they are, the properties of these defects have not changed. They are all shallow acceptors, except AuCdand BiTe, the transition energies of which are far above the threshold of 0.15 eV. While for other defects, it shows that both standard LDA and LDA +Uresults can reflect their defects properties well. For PTeand NTe, the calculated transition energies of (0/–1) by standard LDA and LDA +Uare–0.034/–0.058 eV and 0.06/0.013 eV, respectively. There is not much difference between them with respect to the absolute shallow transition energy level 0.01 eV by LAPW. It can easily be deduced that PTeand NTeare suited to p-type doping. For some extrinsic defects which contain atoms (Cu, Ag, Au, Sb,As) withdelectrons, the results are also improved, although these atoms only occupy ~1.56% in this model.
Above all, there is not much difference between the results of standard LDA and LDA +U, that is, their properties do not change. We also find that the approach of LDA +U, in most instances, is better to reflect the physical properties of the material with d electrons by comparing with standard LDA.
In the following, we will adopt the same way to calculate transition energies of acceptor defect and give the ionization energies of donor defect.

Fig. 1. (Color online) (a) The stable structure is that the Te atom is at the center of the tetrahedron, and (b) a more stable structure is that the anion atom Te and two cation atom Cds are almost in a line.

Fig. 2. (Color online) The acceptor defect (q < 0) transition energy level positions of native defects in CdTe, calculated by LDA, LDA + U and LAPW, respectively.

Fig. 3. (Color online) The donor defect (q > 0) transition energy level positions of native defects in CdTe, calculated by LDA, LDA + U and LAPW, respectively.
From Table 3, we find the native defects CdTe(+2/0) is a shallow level of 0.10 eV by LAPW in the band gap. Standard LDA gives a value of 0.01 eV and LDA +Ushows a value of 0.05 eV, which indicates that LDA +Ugives a better result.However, for other native defects with deep levels in the band gap, such as TeCd(+1/0), the results by different methods are 0.34, 0.345, and 0.625 eV, respectively, indicating that the result of standard LDA can reflect the case better. This conclusion is also true forFrom the data of VTe(+2/0)in Table 3, we find that all the methods show deep donors.LDA + U gives a value of 0.662 eV, because the atomic positions are sensitive to their charge states in negative U systems, such as CdTe,and VTe. It can be explained that charge states influence the transition/ionization energy levelspowerfully, that is, the electrostatic correction value affects the result with charge square.

Table 2. Calculated acceptor defect (q < 0) transition energies of tetrahedron CdTe. The unit of transition energy is eV.

Table 3. Calculated donor defect (q > 0) transition energy levels for tetrahedron native defects. The unit of transition energies is eV.
For extrinsic impurity donor, the calculated (+1/0) transition energy levels of ACd(where A= Al, In, Ga) by LDA + U are at –0.038 eV (above the CBM), 0.347 eV and 0.316 eV (below the CBM). After comparing the results, our calculation suggests that gallium could be a good n-type dopant for CdTe rather than aluminum and indium. For BTe(where B represents I, Br, Cl or F), the calculated transition energy levels at 0.293, 0.341, 0.371, and 0.739 eV are gained by LDA + U,which indicates these defects all have deeper levels. Comparatively speaking, the iodine has the superiority to be as a good n-type dopant in CdTe, while Fluorine is the worst candidate for its very deep level.
4. Conclusion
In this study, the effects of two different calculation methods were investigated. We have systematically employed the methods of standard LDA and LDA + U to calculate the lattice constants, model structures, electronic band structures,the formation energies and transition energies of defects in CdTe, these results were analyzed and compared to the data of LAPW, which derived from other work[9]. The band gap had an obvious improvement by LDA + U because Cd-4d and Te-5p coupling became weaker when appropriate U value was added to Cd atom. Because of its mechanism, the method of DFT gave lower formation energies of the neutral point defects than those obtained from LAPW, and the majority values of LDA + U are closer to LAPW. For transition energies of the charged state defects, except for the intrinsic defects VCdandthe approach of LDA + U were superior to standard LDA to reflect the physical properties of the material with d or f electrons.
After comparing the calculation results of LAPW with standard LDA and LDA + U, we can safely draw a conclusion that the approach of LDA + U not only improves the accuracy from standard LDA method but also reduces the computationally demanding with respect to LAPW method in calculation of defects in CdTe. Therefore, it has a huge potential to model supercells with a large number of atoms and strong electron interactions.
Acknowledgements
The authors gratefully acknowledge the financial support of the National Nature Science Foundation of China (No.51902005) and Young Talent Fund of University Association for Science and Technology in Shaanxi Province of China (No.20180507).
杂志排行

Journal of Semiconductors的其它文章
- S uppression of oxygen and carbon impurity deposition in the thermal system of Czochralski monocrystalline silicon
- A snapback-free and high-speed SOI LIGBT with double trenches and embedded fully NPN structure
- Wireless communication and wireless power transfer system for implantable medical device
- A 0.5–3.0 GHz SP4T RF switch with improved body self-biasing technique in 130-nm SOI CMOS
- Comparative study of various methods for extraction of multiquantum wells Schottky diode parameters
- 4H-SiC trench MOSFET with an integrated Schottky barrier diode and L-shaped P+ shielding region