金属卟啉催化氧化烃类饱和C—H键的理论模拟研究进展
2020-10-20王浩姬东方于艳敏佘远斌
王浩,姬东方,于艳敏,佘远斌
(1 北京工业大学化学化工系,绿色催化与分离北京市重点实验室,北京100124;2 浙江工业大学化学工程学院,绿色化学合成技术国家重点实验室培育基地,浙江杭州310014)
氧化反应是化学工业中的重要反应,通过氧化反应能够制得超过30%的化工产品[1-2]。烃类饱和C—H键的氧化是制备醇和羰基化合物的重要反应,其作为氧化领域的重要组成部分,在化学工业中发挥着重要的作用[3]。但烃类化合物的饱和C—H 键不活泼,氧化反应所需条件苛刻、产物复杂、选择性差[4-5]。如何实现烃类饱和C—H键的选择性氧化一直是化学工业生产中亟待解决的难题。随着绿色化工的进一步推进,采用高活性、高选择性的催化剂实现烃类饱和C—H键的选择性氧化已经成为近年来研究的焦点[6-7]。金属卟啉及金属卟啉衍生物作为细胞色素P450 的核心结构,能够模拟细胞色素P450 在温和条件下活化氧化剂生成卟啉高价金属氧化物[8],并进一步高活性、高选择性地氧化烃类饱和C—H 键,实现烃类饱和C—H 键的功能化[9-12](图1)。金属卟啉作为仿生催化剂在催化氧化烃类饱和C—H键领域已经展现出十分优越的性能,并在工业上具有较好的应用前景。郭灿城教授课题组[13]对金属卟啉仿生催化氧化环己烷制备环己酮的反应进行了研究。与传统的环己烷液相氧化法相比,金属卟啉催化氧化法制备环己酮以极少量的铁卟啉作为仿生催化剂,以空气为氧化剂,环己烷的转化率由4%~5%提高到6.6%,环己酮的选择性由80%左右提高到97.8%。目前金属卟啉仿生催化氧化环己烷已成功实现了工业化生产,推动了金属卟啉仿生催化氧化烃类饱和C—H键的反应由基础研究向工业应用的迈进[14]。甲苯的催化氧化反应在化学工业中也是一类非常重要的反应。金属卟啉作为仿生催化剂催化甲苯氧化可以生成苯甲醇、苯甲醛以及苯甲酸。3种产物的选择性可以通过改变金属卟啉结构来加以调节,能够满足不同化学品工业生产的需要。以四苯基钴卟啉为催化剂,空气为氧化剂,甲苯的转化率能达到9%左右,苯甲醇及苯甲醛的收率为60%[15]。当四苯基锰卟啉催化氧气氧化甲苯时,甲苯的转化率为6%左右,苯甲醇及苯甲醛的收率高达96%[16]。
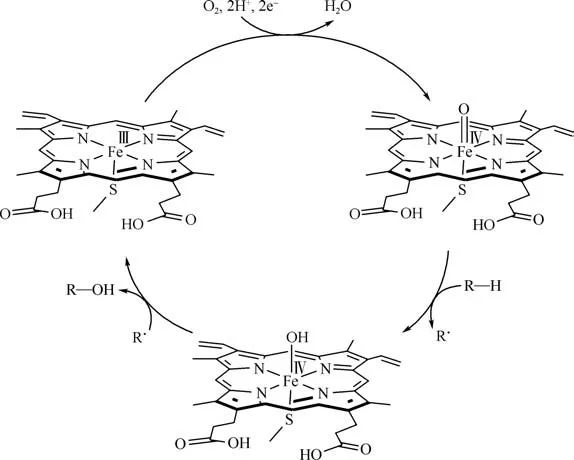
图1 细胞色素P450催化氧化烃类饱和C—H键示意图[12]
由于金属卟啉催化剂的众多优势,近年来,国内外学者对金属卟啉仿生催化剂催化氧化烃类饱和C—H 键的反应进行了大量研究[17]。金属卟啉仿生催化氧化烃类饱和C—H键的研究可以采用实验的方法和理论模拟的方法。采用实验方法研究金属卟啉催化烃类饱和C—H键存在成本较高、对复杂机理难以深入探究等问题。随着计算机运算能力的发展,利用理论模拟方法研究金属卟啉催化氧化烃类饱和C—H键反应的实用性进一步增强。通过理论模拟方法研究金属卟啉催化氧化烃类饱和C—H键过程中各中间体的结构特性,可以探究金属卟啉结构性质与催化活性和选择性之间的关系,得到影响金属卟啉催化活性和选择性的关键因素,为实验上合成高活性高选择性的金属卟啉催化剂提供理论指导。利用理论模拟方法探究金属卟啉催化氧化烃类饱和C—H键的反应机理,可以寻找反应过程的最优路径,更深入地分析和阐明金属卟啉催化氧化烃类饱和C—H键的实验现象和实验结果,并避免实验的盲目性造成人力和原料的浪费。本文对近年来金属卟啉仿生催化氧化烃类饱和C—H键的理论模拟研究进行综述,并对金属卟啉在仿生催化领域的理论模拟前景进行展望。
1 理论模拟方法
随着计算机硬件及软件的快速发展,理论模拟计算在化学化工领域的应用日益成熟。理论模拟研究金属卟啉催化氧化烃类饱和C—H键反应可以采用的理论模拟方法有量子力学(QM)方法、分子力学(MM)方法和量子力学/分子力学(QM/MM)结合方法等。常用的量子力学方法又包括密度泛函理 论(density functional theory,DFT) 方 法[18-19]、从头算(ab initio)方法[20]、半经验(semiempirical)方法[21]等。不同理论模拟方法的特点及适用范围见表1。
量子力学方法借助波函数描述粒子的运动状态,并以薛定谔方程描述波函数的变化规律,用算符或矩阵方法对各种物理量进行计算。对于金属卟啉催化氧化烃类饱和C—H键过程中涉及的各种几何性质、电子结构、反应能量的变化都可以采用量子力学方法进行计算。其中密度泛函理论方法是按Hartree-Fock-Roothaan原理借助变分法或者数值方法,对分子内电子的动能和势能进行计算得到薛定谔方程的近似解——分子波函数。密度泛函理论方法计算精度可通过改变泛函及基组进行调节,操作简便且应用灵活,计算模拟过程中效率较高、误差小、实用性强,目前在金属卟啉体系的计算中颇受关注[22-23]。密度泛函理论方法涉及众多泛函,其中杂化-交换相关泛函B3LYP[24-25]和范围分离泛函ωB97X-D[26]对于卟啉体系计算准确度高,受到众多学者推崇[27-28]。从头算方法不借助任何经验参数,全部严格计算分子积分以求解全电子体系的薛定谔方程。从头算方法计算工作量大,计算代价较高,更适用于小分子结构性质及能量变化的计算。在金属卟啉大环体系的计算中,从头算方法不如密度泛函理论方法计算效率高。半经验方法在计算过程中或忽略一些积分,或在一些积分值上使用经验参数近似求解自洽场分子轨道方程,对计算过程进行了简化,提高了计算效率。但是由于半经验方法计算时引用了近似来简化工作量,对能量计算远不及密度泛函理论方法精确、可靠。目前,半经验方法常用于较大体系结构的初步优化,为进一步高精度计算提供初始结构。当对多系列体系进行计算,比较计算结果,寻求结构与性能的递交规律时,采用半经验方法快速计算也具有重要价值。
分子力学方法运用经典力学模拟分子体系,通过改变分子的几何构型,寻找体系能量最低的结构,在计算分子体系能量与结构方面应用广泛,能够对体系进行构象分析,研究结构与性能之间的关系等。分子力学方法忽略电子的运动,在原子水平上研究体系的结构,适用于研究原子数成千甚至百万的大型体系,计算效率高。但由于分子力学计算过程中需要借助相应的力场参数,导致分子力学方法的使用存在局限性。力场参数不适合或缺少相应的力场参数都会使体系结构和能量的计算不够精确。分子力学方法在金属卟啉体系中可用于结构的构象搜索。
量子力学/分子力学结合方法使用量子力学方法计算体系中重要的、涉及化学键变化的部分,同时利用分子力学方法来计算体系中次要部分的结构和能量。量子力学/分子力学结合方法既包含了量子力学方法的精确性,又利用了分子力学方法的高效性,结合二者的优点,能够快速精确计算体系的相关性质。量子力学/分子力学结合方法在酶活性位点反应、液体或大分子环境中的化学反应机理研究方面应用广泛。在金属卟啉体系的计算研究中,量子力学/分子力学结合方法多用于研究蛋白质环境中金属卟啉参与的催化氧化反应。利用量子力学方法中的密度泛函理论方法计算金属卟啉催化氧化过程的结构性质及能量变化,利用分子力学方法模拟体系的蛋白质环境,以探究蛋白质环境对催化氧化反应的影响。

表1 不同理论模拟方法的特点及适用范围
2 金属卟啉几何及电子性质的理论计算
卟啉是由4个吡咯环的α-碳原子通过4个次甲基桥互联而形成的共轭大环化合物,其母体化合物为卟吩[图2(a)]。卟吩自由碱的氮原子与金属阳离子配位,金属阳离子取代氮原子上的氢原子,生成金属卟啉[图2(b)]。卟啉中心金属离子可以连接不同轴向配体,卟啉环中位和β位碳原子上的氢原子均可被其他基团(中位取代基R1,β 位取代基R2)取代生成金属卟啉衍生物[图2(b)]。金属卟啉结构的变化可以影响其几何性质和电子性质,并进一步影响金属卟啉作为仿生催化剂催化氧化烃类饱和C—H 键的活性和选择性。利用理论模拟的方法,优化金属卟啉几何结构,通过对键长、键角等几何性质,电子密度、前线分子轨道等电子性质的分析,深入挖掘金属卟啉的结构特性,阐明其对催化氧化烃类饱和C—H键的影响。

图2 卟吩及卟啉结构
金属卟啉环外取代基对改变金属卟啉的几何和电子性质起到了重要作用。环外取代基的位置和种类均能影响金属卟啉的几何性质和电子性质。She等[29]利用密度泛函理论方法B3LYP和基组6-31G(d)计算了带有不同环外取代基(—H、—Cl、—NO2、—OH、—OCH3)的四苯基铁卟啉的几何性质。当四苯基铁卟啉中位苯基带有对位取代基时,环外苯基环与卟啉环之间的二面角为64°~69°。当四苯基铁卟啉中位苯基带有邻位取代基时,由于苯基邻位取代基与卟啉环之间存在较大的电子排斥与空间位阻作用,使四苯基铁卟啉环外苯基环几乎垂直于卟啉环。Mobin 等[30]利用密度泛函理论方法B3LYP 和基组6-31G(d)对带有3 种不同环外取代基(苯基、噻吩基、呋喃基)的镁卟啉(图3)结构进行优化,结果发现当环外取代基为苯基和噻吩基时,取代基环平面垂直于镁卟啉环平面。而环外取代基为呋喃基时,呋喃环与镁卟啉环在同一平面上,表明呋喃基和镁卟啉环之间存在更强的共轭相互作用。对于带有不同环外取代基的四苯基钴卟啉,当钴卟啉环外苯基上无取代基或者对位带有一个氯取代基时,钴卟啉为平面大环结构。当钴卟啉环外苯基邻位带有两个氯取代基时,钴卟啉有轻微的扭曲,表现为部分共轭作用。当钴卟啉环外苯基与卟啉环β位的氢原子全部被氯取代时,钴卟啉环弯曲为马鞍型结构。钴卟啉独特的几何性质也反映在Co—N键的平均长度上。马鞍型钴卟啉的Co—N键键长约为1.93Å(1Å=0.1nm),其他构型的钴卟啉Co—N 键键长均约为1.98Å[31]。
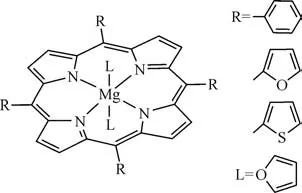
图3 取代镁卟啉的分子结构[30]
除几何性质外,环外取代基对金属卟啉的电子性质也有一定的影响。对不同取代铁卟啉的前线分子轨道进行计算发现[29],四苯基铁卟啉环外带有吸电子取代基(—Cl、—NO2)时可以同时降低其最高占据轨道(HOMO)与最低空轨道(LUMO)的能量,而带有供电子取代基(—OH、—OCH3)时可以提高其HOMO与LUMO的能量。Rosa等[32]利用密度泛函理论方法B3LYP研究铁(Ⅳ)卟啉高价金属氧化物氧化甲烷的过程,在结构优化时对Fe、O原子、轴向配体上的O、H原子及卟啉环上的N原子采用def2-TZVP 基组,其他原子则采用def2-SV(P)基组,在单点能计算时所有原子均采用def2-TZVP基组。结果发现铁(Ⅳ)卟啉高价金属氧化物环外中位吡啶取代基上的正电荷和卟啉环上的正电荷都能减小甲烷电子供体轨道与铁卟啉电子受体轨道之间的能隙,从而降低氢提取过程的反应能垒,提高铁(Ⅳ)卟啉高价金属氧化物的反应活性。Wang等[33]利用密度泛函理论方法B3LYP 和基组LANL2DZ 深入研究了卟啉环外中位供电子取代基和卟啉环外β位侧链对卟啉和锌卟啉(图4)电子性质的影响。借助密度泛函理论计算前线分子轨道能量发现,卟啉环外中位的供电子取代基能够提高卟啉HOMO 的能量。卟啉环外β位侧链中共振能较低的取代基能够降低卟啉及锌卟啉HOMO 和LUMO 的能量以及HOMO与LUMO之间的能隙。

图4 取代卟啉及取代锌卟啉的分子结构[33]
除卟啉环外取代基外,卟啉中心金属离子对金属卟啉几何及电子性质的影响也较为显著。卟啉中心金属离子直接与卟啉大环相互作用。金属离子不同会对金属离子与卟啉环之间的相互作用产生影响,从而改变金属卟啉的几何性质和电子性质。通过对金属卟啉前线分子轨道的计算,发现中心金属离子不同的金属卟啉前线分子轨道组成不同,易变价金属(Fe、Mn、Co)卟啉的前线分子轨道主要成分为金属离子的3d 轨道,不易变价金属(Cu、Zn)卟啉的前线分子轨道为卟啉配体的大π 键[34]。取代四苯基金属卟啉催化氧化环己烷的反应中,卟啉中心金属离子不同,其前线分子轨道能量也有所不同[35]。与钴卟啉和镍卟啉相比,锰卟啉前线分子轨道能隙较低,且锰卟啉前线分子轨道能量与氧气前线分子轨道能量最为接近,锰卟啉活化氧气能力最强。在金属卟啉络合氧气生成卟啉高价金属氧化物后,钴卟啉高价金属氧化物的前线分子轨道能隙最低,且钴卟啉高价金属氧化物的LUMO与环己烷的HOMO 能量接近,有利于电子由环己烷转移至钴卟啉高价金属氧化物,钴卟啉高价金属氧化物氧化底物活性最高。Patra等[36]利用密度泛函理论方法B3LYP 计算了中心金属离子价态不同的铁卟啉结构(图5)。计算过程中对过渡金属铁采用LANL2DZ 赝势基组,其他原子则采用6-31G(d, p)基组。当中心铁离子为正二价时,两个1-甲基咪唑轴向配体咪唑环平面之间的二面角为80.7°,接近相互垂直。当中心铁离子为正三价时,卟啉的结构发生改变,两个1-甲基咪唑轴向配体咪唑环平面之间的二面角为8.6°,二者接近相互平行。

图5 取代铁卟啉的分子结构[36]
卟啉中心金属离子可以连接不同轴向配体,轴向配体的改变也可以影响金属卟啉的性质。She等[29]利用密度泛函理论方法B3LYP和6-31G(d)基组计算了氯轴向配体对四苯基铁卟啉几何性质的影响。氯轴向配体可以增长四苯基铁卟啉中铁离子与吡咯氮原子之间Fe—N键的键长,同时使铁离子由卟啉环外向卟啉环中心移动。对带有不同含氮轴向配体(咪唑及其衍生物、吡啶及其衍生物、哌啶、三乙胺)的锰(Ⅴ)卟啉高价金属氧化物的几何性质进行计算发现,由于含氮轴向配体与中心锰离子的相互作用,影响Mn—N(轴向配体)键键长,从而改变Mn—O 键键长,使其随着Mn—N(轴向配体)键键长减小而增加。其中由于咪唑比吡啶供电子能力更强,咪唑轴向配体与中心锰离子相互作用更强,Mn—N(轴向配体)键键长较小。三乙胺与卟啉间存在较强的空间位阻作用,导致Mn—N(轴向配体)键键长最大[37]。Das等[38]利用密度泛函理论方法B3LYP 及BP86,对Fe、N、O、S 原子采用6-311G(d)基组,其他原子C、H则采用6-31G(d)基组,优化轴向配体为巯基的铁卟啉结构,单点能计算时所有原子采用6-311+G(d)基组计算。研究发现巯基能够与中心铁离子生成较强的共价键,导致铁离子3d 轨道能量增加,减弱铁离子与卟啉环的相互作用,提高Fe—N键的键长。
3 金属卟啉活化氧气
金属卟啉催化氧化烃类饱和C—H 键的反应中,第一个关键步骤是金属卟啉活化氧化剂。目前常用的氧化剂种类众多,如亚碘酰苯(PhIO)[39]、高碘酸钠(NaIO4)[40]、过氧化氢(H2O2)[41]、氧气(O2)[42-43]等。其中由于氧气价格低廉,环境友好,成为金属卟啉催化氧化反应中氧化剂的最佳选择。金属卟啉活化氧气生成卟啉高价金属氧化物,对于金属卟啉催化氧化烃类饱和C—H键的反应具有重要意义[44]。
在众多金属卟啉中,目前研究较多的是铁卟啉对分子氧的活化。Ali 等[45]利用密度泛函理论方法模拟计算了轴向配体为咪唑的铁卟啉与分子氧的反应。模拟计算采用密度泛函理论方法B3LYP,在结构优化时对过渡金属铁采用LANL2DZ赝势基组,其他原子C、H、O、N 则采用6-311G(d,p)基组,在单点能计算时所有原子均采用6-311G(d,p)基组。通过计算研究自旋S=0、1、2、3 时铁卟啉双氧加和物的几何结构及能量,发现铁卟啉双氧加和物自旋S=0,即单重态时最稳定。对于铁卟啉与分子氧之间的反应过程研究发现,两者之间是通过非键相互作用开始的,如电荷-偶极相互作用、偶极-偶极相互作用、范德华相互作用。这些非键相互作用重新分配了铁卟啉的π-电子密度,使铁离子偏离卟啉环平面的距离减小。铁卟啉与分子氧反应的过程中,随着铁卟啉与分子氧逐渐接近,Fe—O距离越来越小。当Fe—O 距离为1.78Å 时,铁离子与卟啉环几乎在同一平面内,铁卟啉与分子氧络合生成稳定的铁卟啉双氧加和物。金属卟啉与分子氧所形成的双氧加和物是研究金属卟啉活化氧气的关键结构。Pierloot 等[46]利用密度泛函理论方法BP86,对比研究了铁卟啉双氧加和物与锰卟啉双氧加和物的几何构型。对过渡金属铁、锰采用ECP-10-MDF赝势基组,其他原子C、H、O、N采用6-31G(d)基组。通过计算不同结合模式的基态结构,即自旋S=0 的单重态铁卟啉双氧加和物及自旋S=3/2 的四重态锰卟啉双氧加和物发现,铁卟啉双氧加和物存在两种end-on构型,见图6(a)和(b)。构型(a)为O—O键在卟啉环上的投影与卟啉环上的Fe—N 键成45°角。构型(b)为O—O键在卟啉环上的投影与卟啉环上的Fe—N 键重合。这两种构型能量相差约2kcal/mol(1kcal=4.18kJ),构型(a)能量更低,结构更稳定。锰卟啉双氧加和物的结构为side-on构型,见图6(c)和(d)。构型(c)为O—O 键在卟啉环上的投影与卟啉环上的Fe—N键成45°角,构型(d)为O—O键在卟啉环上的投影与卟啉环上的Fe—N 键重合,构型(d)为稳定构型。对比计算两种金属卟啉与分子氧反应的结合焓发现,分子氧与锰卟啉的相互作用比与铁卟啉的相互作用强。
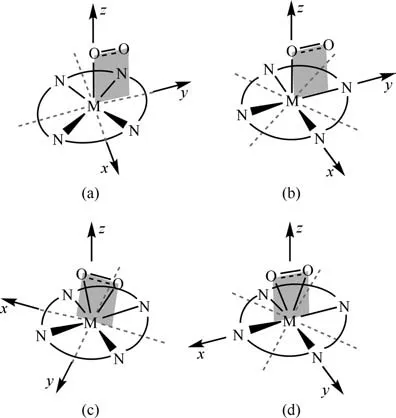
图6 铁卟啉和锰卟啉双氧加和物的几何构型[46]
相比于铁卟啉与分子氧反应所形成双氧加和物的几何结构,铁卟啉双氧加和物的电子结构至今仍存在争议。早在1936 年,Pauling 等[47]通过磁性测量发现铁卟啉双氧加和物的Fe—O键是自旋S=0的单重态铁离子与自旋S=0的单重态分子氧之间的相互作用。1960 年,McClure[48]通过光学吸收光谱、电子自旋共振等实验方法研究提出另一种电子构型,认为Fe—O键是自旋S=1的三重态铁离子与自旋S=1 的三重态分子氧之间的相互作用。之后Goddard等[49]利用从头算方法对Mcclure提出的观点进行了进一步印证。随后在1964 年,Weiss[50]基于铁卟啉双氧加和物合成过程中的光谱数据等提出第三种电子构型,即铁离子与分子氧成键部分为自旋S=1/2 的二重态铁离子与自旋S=1/2 的二重态O2-之间反铁磁耦合。后人在此三种电子构型(图7)的基础上进行了大量研究[27,51-53]。2008 年,Shaik 等[27]利用量子力学/分子力学结合方法对铁卟啉与分子氧的键合性质进行研究,发现铁卟啉与分子氧的键合性质受外界因素的影响,在反应过程中蛋白质环境或轴向配体的微小变化都会影响其电子构型。

图7 铁卟啉双氧加和物的电子构型[53]
除铁卟啉之外,其他金属卟啉活化分子氧的研究近年来也有很大进展。Witko 等[54]对不同金属(锰、钼、钴)卟啉活化分子氧的过程采用密度泛函理论方法GGA-RPBE 和基组DZVP 进行了计算。结果显示锰卟啉和钼卟啉双氧加和物为side-on 构型[图8(a)],其与一个氢原子反应生成氢过氧化物[图8(c)],继而与第二个氢原子反应并脱水最终生成卟啉高价金属氧化物[图8(d)]。钴卟啉双氧加和物为end-on 构型[图8(b)],与两个氢原子反应后,不易脱去水分子,所以不会生成最终的卟啉高价金属氧化物。Witko 等[55]又采用密度泛函理论方法LDA-VWN 及GGA-RPBE,基组DZVP 对锰卟啉活化分子氧的热力学性质进行计算。分子氧与锰卟啉反应为吸热过程,分子氧与还原的锰卟啉反应为放热过程。所以分子氧更倾向于在锰卟啉还原后与其反应生成锰卟啉双氧加和物,最终O—O键断裂生成锰(Ⅴ)卟啉高价金属氧化物。对于四苯基钴卟啉与分子氧的反应,研究发现四苯基钴卟啉与分子氧结合后,四苯基钴卟啉结构发生了明显变化。四苯基钴卟啉原为平面环状结构,中位苯基环垂直于卟啉环平面。与分子氧结合后,四苯基钴卟啉平面结构轻微弯曲为马鞍形结构,环外苯基环旋转与四苯基钴卟啉平面形成74.5°二面角。立体旋转促进电子由环外苯基向钴卟啉大环转移,进而转移至分子氧,有利于分子氧的活化[56]。

图8 金属卟啉活化分子氧过程中的不同构型[54]
金属卟啉活化分子氧的反应受金属卟啉几何性质的影响。铁卟啉双氧加和物中,含氮轴向配体(氨、咪唑、吡啶及其衍生物)能够吸引铁离子偏离卟啉环,形成一个空腔,促进铁卟啉与分子氧结合。轴向配体上与中心铁离子相连的氮原子的电子密度越大,铁卟啉与分子氧反应活性越强。生成铁卟啉双氧加和物后,分子氧上的负电荷越多,其活性越强,促进其参与后续的催化氧化反应[57]。Boyd等[58]借助密度泛函理论方法ωB97X-D,对过渡金属铁采用6-311+G(f)基组,对N、S、O 原子采用6-311+G(d)基组,对C、H 原子采用6-31G 基组,计算研究轴向配体对铁卟啉活化分子氧的影响。研究发现,除轴向配体上具有较高的电子密度外,轴向配体与铁卟啉之间较强的相互作用也是保障铁卟啉与分子氧具有较强亲和力的必要因素。在带有不同环外取代基的四苯基金属卟啉活化分子氧过程中,当四苯基金属卟啉环外中位苯基对位为吸电子取代基时,四苯基金属卟啉环上的负电荷减少。吸电子取代基越强,越容易降低卟啉环上的负电荷密度,增强分子氧与中心金属离子的结合能力,从而有利于金属卟啉活化分子氧[36,59]。除金属卟啉轴向配体与环外取代基之外,金属卟啉大环结构的差异也会影响其与分子氧的反应。几种卟啉大环弯曲程度不同的四苯基钴卟啉与分子氧反应,卟啉大环弯曲程度较大的马鞍形结构钴卟啉在与分子氧结合过程中存在空间位阻,不利于分子氧的活化,当钴卟啉大环为平面时,活化分子氧的能力最强[31]。
4 卟啉高价金属氧化物氧化烃类饱和C—H键
卟啉高价金属氧化物作为金属卟啉催化氧化烃类饱和C—H键的活性物种,能够在温和的条件下氧化烃类饱和C—H 键[12]。卟啉高价金属氧化物氧化烃类饱和C—H键的反应机理,一直是仿生催化领域研究的热点。对反应机理的深入剖析有助于探究其催化剂本质,从而为进一步设计新型高效的金属卟啉催化剂提供方向。目前,在卟啉高价金属氧化物氧化烃类饱和C—H键的反应中,采用理论模拟方法研究较多的是铁卟啉和锰卟啉的高价金属氧化物对烃类饱和C—H键的氧化。

图9 铁卟啉催化氧化烃类饱和C—H键的反弹机理[63-64]
Groves 等[60-63]提出的反弹机理目前广泛应用于铁卟啉催化氧化烃类饱和C—H键的反应中。反弹机理涉及两个关键步骤,首先铁(Ⅳ)卟啉高价金属氧化物作为活性物种从烷烃饱和C—H键提取氢原子,形成高价羟基取代铁(Ⅳ)卟啉及烷基自由基,然后羟基反弹至烷基自由基生成最终的羟基化产物,见图9[63-64]。其中卟啉高价金属氧化物从烷烃饱和C—H 键提取氢原子的过程为反应的速控步骤。基于反弹机理,Shaik等[65-67]利用密度泛函理论方法B3LYP,对过渡金属铁采用LACVP 基组,其他原子采用6-31G基组,研究P450活化甲烷C—H键的反应,并在此基础上提出了两态反应机理。两态反应机理中,铁(Ⅳ)卟啉高价金属氧化物作为氧化的活性中心,自旋S=1/2 时为低自旋(low spin,LS)二重态,自旋S=3/2 时为高自旋(high spin,HS)四重态。两种自旋态的反应路径都是铁(Ⅳ)卟啉高价金属氧化物从烷烃中提取氢原子,然后羟基反弹至烷基自由基生成醇。当铁(Ⅳ)卟啉高价金属氧化物为四重态时,羟基反弹步骤存在一个较小的能垒,为分步反应。当铁(Ⅳ)卟啉高价金属氧化物为二重态时,羟基直接反弹至烷基自由基生成羟基化产物,为协同反应。整个反应过程中两种自旋态能量相差较小,二者共同作用使烷烃羟基化反应得以进行,见图10。对不同自旋态下铁(Ⅳ)卟啉高价金属氧化物夺氢过程的过渡态结构进行分析发现,当铁(Ⅳ)卟啉高价金属氧化物为二重态及四重态时,过渡态结构中Fe—O—H 夹角约为120°[图11(a)]。当铁(Ⅳ)卟啉高价金属氧化物为能量更高的六重态时,过渡态结构中Fe—O—H夹角约为180°[图11(b)][68]。基于两态反应机理,国内外学者对铁(Ⅳ)卟啉高价金属氧化物氧化烷烃饱和C—H键的反应进行了诸多研究[69-74]。Wu等[73]利用密度泛函理论方法B3LYP和基组6-31G对铁(Ⅳ)卟啉高价金属氧化物氧化甲烷的过程进行模拟发现,在甲烷与铁(Ⅳ)卟啉高价金属氧化物不断接近的初期,甲烷C—H 键键长基本不变,甲烷与铁(Ⅳ)卟啉高价金属氧化物之间的相互作用是能量变化的决定因素。当两者接近至C—O 距离为2.59~2.60Å 时,C—O 距离保持不变,氢原子不断向氧原子移动,C—H 键键长增长,直至生成过渡态结构,此时铁(Ⅳ)卟啉高价金属氧化物的电子结构及甲烷C—H键的解离能是能量变化的决定因素。研究轴向配体对铁(Ⅳ)卟啉高价金属氧化物氧化环己烷饱和C—H键的影响发现,供电子轴向配体能够加强FeO—H键,削弱Fe=O 键,从而提高铁(Ⅳ)卟啉高价金属氧化物提取环己烷氢原子及羟基反弹的反应活性[74]。
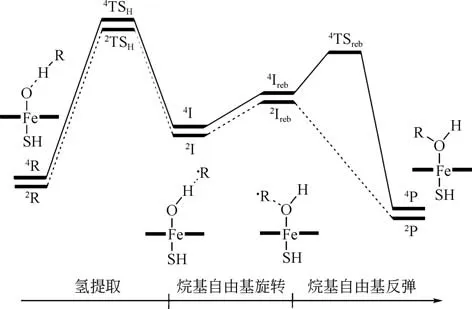
图10 铁卟啉催化氧化烷烃羟基化反应的两态反应机理[67]

图11 不同自旋态的铁(Ⅳ)卟啉高价金属氧化物氧化烷烃C—H键的过渡态结构[68]
铁(Ⅳ)卟啉高价金属氧化物氧化烷烃C—H 键的反应能垒主要受烷烃C—H键解离能的影响,烷烃C—H 键解离能越大,反应能垒越高[75],但外围环境的影响也可能改变反应能垒的大小。Wang等[76]借助密度泛函理论方法B3LYP 计算铁(Ⅳ)卟啉高价金属氧化物连续两次羟基化异戊烷的反应。结构优化时对过渡金属铁采用LACVP 基组,其他原子采用6-31G 基组,即LACVP/6-31G,计算单点能时采用更高精度基组LACVP+(d,p)/6-311+G(d,p)。两次羟基化反应都遵循两态反应机理。第二次羟基化过程中,由于底物上羟基与铁(Ⅳ)卟啉高价金属氧化物的氧原子之间存在氢键作用,反应能垒比第一次羟基化反应能垒高2kcal/mol。铁(Ⅳ)卟啉高价金属氧化物氧化莰酮饱和C—H 键过程中,当铁(Ⅳ)卟啉高价金属氧化物活性位点存在水分子时,水分子与铁(Ⅳ)卟啉高价金属氧化物轴向氧原子之间存在氢键作用,由于过渡态结构中电荷由莰酮转移至氧原子,增加了氧原子的负电荷,致使过渡态结构的氢键作用更强,降低了氢提取步能垒[77]。对于反应环境的影响,可以采用量子力学/分子力学结合方法计算。量子力学/分子力学结合方法可以用分子力学方法计算环境中的物质与铁(Ⅳ)卟啉高价金属氧化物之间的相互作用,用量子力学方法计算铁卟啉对烃类分子的催化氧化[78-81]。Shaik等[79]利用量子力学/分子力学结合计算方法对铁(Ⅳ)卟啉高价金属氧化物氧化小分子烷烃(甲烷、乙烷、丙烷)的反应过程进行研究发现,小分子烷烃容易逃离铁(Ⅳ)卟啉高价金属氧化物的活性位点,全氟癸酸的存在可以使小分子烷烃固定在活性位点附近,从而提高铁(Ⅳ)卟啉高价金属氧化物对小分子烷烃的氧化。蛋白质环境与铁(Ⅳ)卟啉高价金属氧化物之间存在非键相互作用(静电相互作用、范德华相互作用、极化作用),对铁(Ⅳ)卟啉高价金属氧化物的反应活性也有一定影响[80]。Lu等[81]借助量子力学/分子力学结合方法计算了Thr303 残基对核心结构为铁(Ⅳ)卟啉高价金属氧化物的细胞色素P450氧化乙醇的影响。当不存在Thr303 残基时,P450 既可以提取乙醇饱和C—H键的氢原子,也可以提取乙醇羟基上的氢原子,两种反应路径相互竞争。当反应环境中加入Thr303 残基后,Thr303 残基与乙醇之间形成氢键,使乙醇分子以特定构型固定于活性位点附近,促使P450提取乙醇羟基上的氢原子,导致P450活化氧化乙醇饱和C—H的能力降低。
除铁(Ⅳ)卟啉高价金属氧化物外,锰(Ⅴ)卟啉高价金属氧化物氧化烷烃C—H键同样遵循反弹机理。对于轴向配体为咪唑、吡啶、2,6-二甲基吡啶的四苯基锰(Ⅴ)卟啉高价金属氧化物和五氟苯基锰(Ⅴ)卟啉高价金属氧化物,Rezaeifard 等[82]借助密度泛函理论方法B3LYP 计算其氧化环己烷过程中氢提取步过渡态的结构及能量。计算过程中对过渡金属锰采用LANL2DZ 赝势基组,其他原子采用6-31G(d)基组。由于配体的供电子能力不同,导致带有不同轴向配体的四苯基锰(Ⅴ)卟啉高价金属氧化物的氧化能力大小顺序为咪唑>吡啶>2,6-二甲基吡啶。但对于五氟苯基锰(Ⅴ)卟啉高价金属氧化物,由于吡啶环上的氢原子、2,6-二甲基吡啶中甲基上的氢原子与卟啉环外苯基的氟原子之间存在氢键作用,显著降低了氢提取步能垒,导致带有不同轴向配体的五氟苯基锰(Ⅴ)卟啉高价金属氧化物的氧化能力为吡啶>2,6-二甲基吡啶>咪唑。除了氢提取步能垒之外,羟基反弹步能垒也受轴向配体的影响。Groves 等[83]借助密度泛函理论方法B3LYP,对过渡金属锰采用LACVP(d,p)基组,其他原子采用6-31G(d,p)基组,计算研究轴向配体为H2O、CH3CN、F-、OH-、O2-的锰卟啉的羟基反弹步能垒,通过研究锰卟啉与甲烷反应的羟基反弹步势能曲线(图12)发现,当轴向配体为F-、OH-、O2-时,羟基反弹能垒显著增加。

图12 锰(Ⅳ)卟啉氢氧化物与甲基自由基反应的反弹势能曲线[83]
除烷烃中的饱和C—H 键外,金属卟啉也能催化氧化烯烃分子中的饱和C—H 键。Harvey 等[84]利用量子力学/分子力学结合方法计算了P450氧化丙烯、环己烯饱和C—H键的选择性,发现P450对特定活性位点的相对反应活性是影响反应选择性的关键因素。由于烯烃结构中存在饱和C—H键和C=C键两种可氧化位点。在金属卟啉催化氧化烯烃的反应中,金属卟啉可以选择性催化氧化饱和C—H键发生羟基化反应生成相应的醇,也可以选择性催化氧化较为活泼的C=C 键生成环氧化物[85]。Sharma等[86]借助密度泛函理论方法B3LYP,对过渡金属铁采用LACVP 基组,其他原子采用6-31G 基组,计算探究了铁(Ⅳ)卟啉高价金属氧化物催化氧化丙烯体系中羟基化和环氧化这两个竞争反应的机理(图13)。丙烯可以通过烯丙位氢原子或不饱和碳原子与铁(Ⅳ)卟啉高价金属氧化物发生反应。丙烯烯丙位氢原子与铁(Ⅳ)卟啉高价金属氧化物反应时,遵循反弹机理,发生羟基化反应。丙烯不饱和碳原子与铁(Ⅳ)卟啉高价金属氧化物反应,Fe—O键断裂,C—O键生成,最终环闭合生成环氧化物。两个竞争反应的存在导致烯烃的催化氧化产物选择性差,产物复杂。Gupta 等[87]运用密度泛函理论方法B3LYP计算铁(Ⅳ)卟啉高价金属氧化物氧化环己烯的反应。结构优化时对过渡金属铁采用LACVP基组,其他原子采用6-31G 基组,即LACVP/6-31G。计算单点能时采用更高精度基组LACV3P/6-311+G(d)。结果发现环己烯C=C 环氧化过程能垒较低,为主要反应。但是由于环己烯羟基化过程的能垒仅比环氧化过程高3.2kcal/mol,使通过改变外部条件来转换反应路径,提高反应对羟基化产物的选择性成为可能。当环己烯上的氢原子被氘原子取代后,由于存在隧穿效应,当温度降低时,C—H羟基化过程成为优选路径。

图13 铁(Ⅳ)卟啉高价金属氧化物氧化丙烯的两种反应路径[86]
金属卟啉催化氧化芳烃α-C—H键可以生成芳醇。对于锰(Ⅴ)卟啉高价金属氧化物氧化甲苯的反应,研究发现锰(Ⅴ)卟啉高价金属氧化物上氧原子的自由基特征是影响其活性的重要因素[88]。此外,锰(Ⅴ)卟啉高价金属氧化物的轴向配体能够影响其氧化甲苯反应的活性,对于轴向配体为H2O、OH-、O2-的锰(Ⅴ)卟啉高价金属氧化物,其氧化甲苯的反应活性为H2O>OH->O2-[89]。卟啉的立体结构也能够影响反应的选择性。研究手性铁卟啉(图14)催化氧化乙苯反应的选择性发现,反应生成的1-苯乙醇存在两种对映异构体:(R)-1-苯乙醇、(S)-1-苯乙醇。氢提取步骤的过渡态结构中,乙苯与手性铁卟啉之间的相互作用是控制立体选择性的主要因素。由于生成(R)-1-苯乙醇的过渡态结构中,乙苯的苯基和铁(Ⅳ)卟啉高价金属氧化物的环外取代基萘基之间存在π-π堆积作用,所以生成(R)-1-苯乙醇的能垒更低,(R)-1-苯乙醇为反应的主产物[90]。卟啉高价金属氧化物氧化苯烃α-C—H键生成芳醇的同时,也能催化氧化苯环上的C—H键生成相应的酚和酮[91]。铁卟啉(Ⅳ)高价金属氧化物氧化甲苯的反应中,甲苯α-C—H键氧化生成苯甲醇的反应能垒较低,为主要反应。苯环上的C—H键被氧化生成的对甲基甲酚和对甲基环己酮为副产物。但铁(Ⅳ)卟啉高价金属氧化物轴向巯基配体与蛋白质环境之间的氢键相互作用会对反应路径产生影响,使甲苯α-C—H键羟基化过程反应能垒增大,苯环上C—H 键氧化过程能垒减小[92]。此外,同位素效应也会影响反应过程。对于铁(Ⅳ)卟啉高价金属氧化物氧化乙苯的反应,De Visser[93]利用非限制性密度泛函理论方法UB3LYP,对过渡金属铁采用LACVP基组,其他原子采用6-31G基组,计算发现当乙苯上所有氢原子被氘原子取代后,苯环上C—H键因氧化反应能垒降低而变得更具有竞争性。

图14 手性双萘酚铁卟啉的分子结构[90]
5 结语
理论模拟计算在研究金属卟啉仿生催化氧化烃类饱和C—H键领域得到了广泛应用,并在研究金属卟啉的结构性质及催化氧化机理等方面取得了重要的进展,为金属卟啉催化氧化烃类饱和C—H键的应用提供了理论指导。在理论模拟方法方面,应用更高精度的量子力学方法计算金属卟啉的几何结构和电子结构,以及充分发挥不同方法的优势,扬长避短,有机结合多种方法,用于金属卟啉体系及其外围环境的计算是今后理论模拟研究金属卟啉仿生催化体系的发展趋势。在研究内容方面,虽然金属卟啉催化氧化烃类饱和C—H键的机理研究已取得重大进展,但是由于各种机理的复杂性,机理的探究仍然是具有重要价值的研究课题。此外,金属卟啉结构的改变能够影响金属卟啉的催化活性及选择性。其中金属卟啉催化氧化反应的选择性与其结构之间的关系具有较大的研究空间。通过改变金属卟啉结构来调节其催化氧化烃类底物的选择性,能够更加灵活地满足制备各种化学品的需求,具有较大的工业价值。金属卟啉催化领域的研究将更倾向于实验探究与理论模拟相结合的模式。理论模拟为实验提供指导,并深入解释实验现象,同时实验进一步验证理论模拟结果,二者相辅相成,紧密结合,共同推动金属卟啉仿生催化领域的发展。
