Role of succinate dehydrogenase deficiency and oncometabolites in gastrointestinal stromal tumors
2020-10-09YueZhaoFeiFengQingHongGuoYuPingWangRuiZhao
Yue Zhao, Fei Feng, Qing-Hong Guo, Yu-Ping Wang, Rui Zhao
Abstract Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract. At the molecular level, GISTs can be categorized into two groups based on the causative oncogenic mutations. Approximately 85% of GISTs are caused by gain-of-function mutations in the tyrosine kinase receptor KIT or platelet-derived growth factor receptor alpha (PDGFRA). The remaining GISTs, referred to as wild-type (WT) GISTs, are often deficient in succinate dehydrogenase complex (SDH), a key metabolic enzyme complex in the tricarboxylic acid (TCA) cycle and electron transport chain. SDH deficiency leads to the accumulation of succinate, a metabolite produced by the TCA cycle. Succinate inhibits α-ketoglutarate-dependent dioxygenase family enzymes, which comprise approximately 60 members and regulate key aspects of tumorigenesis such as DNA and histone demethylation, hypoxia responses, and m6A mRNA modification. For this reason, succinate and metabolites with similar structures, such as D-2-hydroxyglutarate and fumarate, are considered oncometabolites. In this article, we review recent advances in the understanding of how metabolic enzyme mutations and oncometabolites drive human cancer with an emphasis on SDH mutations and succinate in WT GISTs.
Key Words: Gastrointestinal stromal tumors; Succinate dehydrogenase; Oncometabolite; Succinate; α-ketoglutarate-dependent dioxygenase; Epigenetics
INTRODUCTION
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal (GI) tract. At the molecular level, GISTs can be categorized into two groups based on the causative oncogenic mutations. The major group, which accounts for approximately 85% of total GISTs and mainly occurs in adults, is caused by gain-of-function mutations in the receptor tyrosine kinase (RTK) KIT or plateletderived growth factor receptor alpha (PDGFRA)[1,2]. The remaining 15% of GISTs, known as wild-type (WT) GISTs, do not contain mutations in RTKs[1-3]. Consequently, tyrosine kinase inhibitors such as imatinib, which have been effective in treating GISTs with RTK mutations[4,5], show little benefit in patients with WT GISTs[6-8]. Surgery remains the primary and recommended treatment of nonmetastatic WT GISTs[9,10].
In 2011, Janewayet al[11]reported that deficiency of the succinate dehydrogenase complex (SDH), either caused by loss-of-function mutations or reduced expression of genes encoding SDH, was associated with most WT GISTs. This discovery was soon confirmed by multiple independent studies[12-19]. Similar to those of other gastric tumors, the clinical manifestations of SDH-deficient GISTs include bleeding and discomfort in the GI tract[20]. In a molecular genetic study that analyzed more than 1100 patient samples, the patients with SDH-deficient GISTs were most frequently children and young adults (approximately 85%-90% of patients) and were more frequently in female patients with a predilection ratio over 2:1[14]. Furthermore, SDH-deficient GISTs occur primarily in the stomach, particularly in the distal stomach and antrum[14].
SDH, also known as succinate-coenzyme Q reductase or mitochondrial complex II, is a key metabolic enzyme complex in the tricarboxylic acid (TCA) cycle and electron transport chain. SDH oxidizes succinate to fumarate, which is a key step of the TCA cycle that regulates carbon metabolism and energy production in the cells (Figure 1). SDH deficiency changes cellular metabolism, such as the demand for extracellular pyruvate and the biosynthesis of aspartate[21,22]. Metabolic reprogramming in cancer cells has long been noticed but is primarily considered the consequence of tumorigenesis[23-27]. Emerging evidence, however, has demonstrated that metabolic changes caused by mutations in metabolic enzymes, such asSDHmutations in GISTs, serve as the driver of human cancer. Additional oncogenic mutations in metabolic enzymes include isocitrate dehydrogenase (IDH1orIDH2) mutations in low-grade gliomas (LGGs) and acute myeloid leukemia (AML) and fumarate hydratase (FH) mutations in hereditary leiomyomatosis and renal cell cancer[28-35]. How these metabolic enzyme mutations drive tumorigenesis is only partially understood. It has been proposed that these mutations can distort mitochondria-mediated apoptosis and activate the hypoxia response pathway. In recent years, emerging evidence indicates that these mutations accumulate metabolites affecting the α-ketoglutarate (α-KG)-dependent dioxygenase family enzymes, which regulate key aspects of tumorigenesis, such as epigenetic modification, hypoxia responses, and RNA methylation (Figure 2). In this article, we review recent advances in understanding how metabolic enzyme mutations and oncometabolites drive human cancer with an emphasis onSDHdeficiency and succinate in WT GISTs.

Figure 1 Mutations in metabolic enzymes produce oncometabolites. Shown are genetic mutations in tricarboxylic acid (TCA) cycle enzymes (underscored) involved in generating oncometabolites (bold). Isocitrate dehydrogenase (IDH) mutations are neomorphic, producing proteins with the modified function of producing D-2-hydroxyglutarate (D-2HG), while succinate dehydrogenase (SDH) and fumarate hydrase (FH) mutations are loss-of-function mutations that lead to the accumulation of succinate and fumarate, respectively. α-KG: α-ketoglutarate.
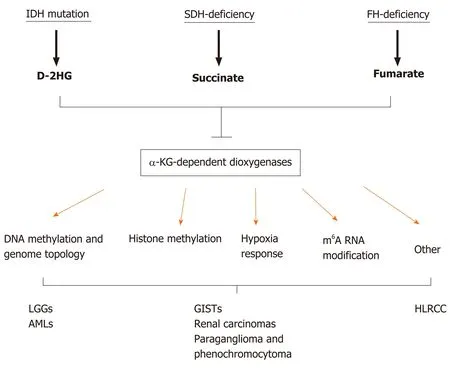
Figure 2 Metabolic enzyme mutations lead to the accumulation of oncometabolites, which competitively inhibit α-ketoglutaratedependent dioxygenases. α-KG: α-ketoglutarate; AMLs: Acute myeloid leukemias; D-2HG: D-2-hydroxyglutarate; FH: Fumarate hydrase; HLRCC: Hereditary leiomyomatosis and renal cell cancer; IDH: Isocitrate dehydrogenase; LGGs: Low-grade gliomas; SDH: Succinate dehydrogenase.
SDH DEFICIENCY, ONCOMETABOLITES, AND GISTS
SDH is a key component of both the TCA cycle and the electron transport chain (ETC). Localized in the inner membrane of mitochondria, the SDH holoenzyme consists of four subunits, SDHA, SDHB, SDHC, and SDHD, and two assembly factors, SDHF1 and SDHF2[36]. Among the four subunits, SDHA catalyzes succinate to fumarate in the TCA cycle. SDHB is involved in the oxidation of ubiquinone to ubiquinol in the ETC, while SDHC and SDHD are mainly responsible for anchoring the SDH protein complex to mitochondria. Loss-of-function mutation in any of the four subunits destabilizes the SDH protein complex and eliminates the entire SDH enzymatic activity. Mutations in all SDH subunits have been identified in GISTs as well as several other human cancers such as rental carcinoma, leukemia, and familial paraganglioma and pheochromocytoma[37-42]. Among the SDH subunits, mutations inSDHAare most frequent, accounting for approximately 30% of total SDH-deficient GISTs[12-19,43]. Notably, approximately 50% of SDH-deficient GISTs are not caused by genetic mutations in any of the SDH subunits. Instead, SDH deficiency in these GISTs results from a lack of expression of the SDH enzyme complex, presumably by mutations elsewhere that affect the expression or turnover of the SDH subunits[15,20].
The loss of SDH enzymatic activity by a loss-of-function mutation or a lack of gene expression leads to the accumulation of succinate[44,45], a metabolite produced from the TCA cycle (Figure 1). Under normal conditions, SDH rapidly converts succinate into fumarate by passing two protons to ubiquinone to initiate the ETC, which is the major process to generate the energy-carrying molecule adenosine triphosphate (ATP). This process is disrupted in SDH-deficient cells. The blockage of succinate conversion to fumarate leads to consequences beyond simply affecting the efficiency of the TCA cycle and the ETC. To adapt to the disruption of the TCA cycle, cells must rewire cellular metabolism by initiating compensation pathways. For example, SDH-deficient cells increase activities in glycolysis, lactate production, and pentose phosphate pathways[46]. More importantly, succinate also functions as a competitive inhibitor of α-KG, which is not only a metabolite in the TCA cycle for energy metabolism but also a co-factor required by the α-KG-dependent dioxygenases. α-KG-dependent dioxygenases catalyze hydroxylation reactions on biomolecule substrates, including DNA, RNA, protein, and lipids[47,48]. Members of the α-KG-dependent dioxygenase family include DNA hydroxylases, histone demethylases, RNA demethylases, and prolyl hydroxylases, which regulate cellular processes such as the demethylation of DNA, histone and nonhistone proteins, and RNA molecules and the responses to hypoxic conditions (Figure 2)[49,50]. Dysregulation of these processes has been considered the driving force of human cancers[51,52]. Because of this tumor-promoting role, succinate together with D-2-hydroxyglutarate (D-2HG) and fumarate, which are produced byIDHandFHmutations, respectively, are dubbed oncometabolites[53].
ONCOMETABOLITES AND EPIGENETICS
Mutations in key metabolic enzymes invariably alter the composition and concentration of metabolites in cells. Generally, there are two nonexclusive ways that metabolites can epigenetically reprogram the affected cells. First, changes in the abundance of metabolites such as acetyl-CoA and S-adenosyl methionine (SAM), which are substrates for key biochemical reactions such as acetylation and methylation, can affect the epigenetic status of the entire genome. Second, the accumulation of oncometabolites can affect the activities of α-KG-dependent dioxygenases, which are involved in the regulation of specific epigenetic modifications and related biological pathways.
Metabolites as substrates for key epigenetic modification reactions
Acetylation and methylation of histone proteins and methylation of genomic DNA are the major modifications that shape the epigenetic landscape of cells. Acetylation involves covalently linking an acetyl group to the Nεamino group of lysine residues of histone and nonhistone proteins. By adding an acetyl group, acetylation neutralizes the positively charged lysine residue, which often causes changes in the enzymatic activity, localization, stability, and interaction profiles of the modified proteins[54-56]. More than 8000 unique acetylation sites have been identified in histone and nonhistone proteins in mammalian cells[57-59]. Histone acetylation, catalyzed by histone acetyltransferases, is associated with genomic regions with active transcription[54,60,61]. As acetyl-CoA is the sole acetyl group donor for protein acetylation, its abundance is one of the key determinants of acetylation reactions. There are three major pathways to produce acetyl-CoA: Pyruvate oxidation, β-oxidation of fatty acids, and ATP-citrate lyase-mediated production from citrate[62]. As a result, cells with active acetyl-CoA production are often associated with increased levels of histone acetylation and gene transcription[63].
Methylation can occur on both protein and genomic DNA, and both forms of modification utilize SAM as the primary donor of the methyl group[64]. Histone methylation involves the covalent addition of one to three methyl groups onto lysine or arginine residues of N-terminal tails by protein methyltransferases, while DNA methylation involves covalent modification of cytosine (C) residues by DNA methyltransferases (DNMTs). In contrast to histone acetylation, which is always associated with active transcription, histone methylation could impact transcription positively or negatively, depending on the specific lysine residues that are modified. For example, histone H3K9 methylation is associated with heterochromatin structures with little transcriptional activity, while histone H3K4 methylation often indicates actively transcribed promoters or enhancers[65]. DNA methylation, particularly within the promoter regions, is associated with transcriptional silencing. Biosynthesis of SAM involves the conjugation of an ATP molecule to homocysteine by methionine adenosyltransferases. The cellular abundance of SAM, presumablyviamodulating DNA and histone methylation modifications, has been suggested to regulate the proliferation and viability of liver cancer cells[66-68].
Metabolites as regulators of epigenetic modifying enzymes
Metabolites can regulate epigenetic modifying enzymes in two mechanisms. First, epigenetic modifying enzymes can be directly methylated or acetylated, which leads to changes in protein conformation, localization, and enzymatic activities. For example, p300, a histone acetyltransferase implicated in several human cancers, including gastric carcinomas[69-72], activates its catalytic activity by autoacetylation[73].
In the second mechanism, metabolites serve as cofactors or antagonists of epigenetic modifying enzymes. The best-known example is α-KG and the related oncometabolites (i.e., succinate, D-2HG, and fumarate) in the regulation of the α-KG-dependent dioxygenase family[74]. Mutations in metabolic enzymes such as SDH, IDH, and FH lead to excessive accumulation of succinate, D-2HG, and fumarate, respectively, all of which share structural similarity with α-KG and competitively inhibit α-KGdependent dioxygenases (Figure 2)[75-79]. Such inhibition affects epigenetic processes, including DNA and histone demethylation, responses to hypoxia, and other biological processes, such as RNA demethylation and DNA damage repair.
Oncometabolites that regulate DNA methylation and genome topology:D-2HG produced byIDHmutations in LGGs and AMLs is the best-studied oncometabolite. Long before the discovery ofIDHmutations as the genetic drivers of LGGs, the genome of a subset of brain tumors was noted to be hypermethylated, referred to as the glioma CpG island methylator phenotype[80]. Subsequent genetic studies associated this group of gliomas with brain tumors harboring neomorphic mutations in theIDH1orIDH2genes[33,81-83]. Under normal conditions, IDH1 and IDH2 convert isocitrate into α-KG (Figure 1). In LGG cells, neomorphicIDHmutations acquire the novel function to further oxidize α-KG into D-2HG, which competitively inhibits all α-KG dependent dioxygenases including the TET-family DNA methylcytosine hydroxylases (i.e., TET1, TET2, and TET3)[75,84]. The methylation status of the genome is determined by the dynamic balance between DNA methylation by DNMTs and demethylation by TETs. The TET enzymes convert 5-methylcytosine to 5-hydroxymethylcytosine[85], which is the first step in the active DNA demethylation pathway[86]. Gain-of-function mutations in DNMTs and loss-of-function mutations in the TET family enzymes lead to genome hypermethylation and have been identified in malignancies such as leukemia, lymphoma, melanoma, and colorectal cancer[87]. Therefore, the hypermethylation phenotype likely contributes to the tumorigenicity of LGGs. SDH-deficient GISTs and FH-deficient tumors similarly exhibit a genome-wide hypermethylated phenotype because of inhibition of the TET family enzymes[11,78,88,89]. Methylation on the promoter of the O6-methylguanine DNA methyltransferase (MGMT) gene has been a biomarker for the efficacy of alkylating agents in LGGs, colorectal carcinomas, and several other cancers[90-92]. Recently, methylation of theMGMTpromoter has been detected in approximately two-thirds of SDH-deficient GISTs, which is significantly more frequent than the SDH-proficient counterparts[93]. However, whether theMGMTpromoter methylation status can similarly predict the response of SDH-deficient GISTs to alkylating agents remains to be investigated.
DNA methylation usually decreases transcription activity by reducing the binding of methylation-sensitive transcription factors and/or by recruiting additional chromatin modification enzymes to create a nonpermissive chromatin environment[94]. The altered transcriptional profile may change cellular physiology that favors tumorigenesis. For example, SDH-deficient tumors with DNA hypermethylation are more invasive than SDH-WT tumors, which is at least partially due to transcriptional dysregulation of genes associated with epithelial-to-mesenchymal transition[88,95].
Very recently, DNA methylation has been demonstrated to regulate transcription by altering genomic topology. This primarily works through CTCF, a DNA-binding insulator protein that only binds to unmethylated DNA recognition motifs[96,97]. CTCF forms protein complexes with cohesins that define chromatin boundaries and establish topologically associated domains[98-100]. One of the key functions of insulators is to prevent promiscuous interactions between enhancer elements and promoters of oncogenes[101]. InIDH-mutant LGG cells, CTCF fails to bind a fraction of putative binding sites, which leads to altered chromatin topology and aberrant expression of associated genes[102]. Gain-of-function mutations inPDGFRAare among the most common driver mutations of glioblastomas (GBMs) but are rare in LGGs[103-105]. CTCF failing to bind to recognition motifs adjacent to thePDGFRApromoter inIDH-mutant LGG cells allows interaction between thePDGFRApromoter and a nearby enhancer element, which leads to increased expression ofPDGFRAin LGG tumor cells[102]. Interestingly, treating LGG cells with 5-azacytidine, an FDA-approved DNAdemethylating agent[106], not only partially restores CTCF binding but also reducesPDGFRAexpression, suggesting that modulating DNA methylation status andPDGFRAexpression could be a potential strategy to treatIDH-mutant LGGs[102].
Similarly, DNA hypermethylation in SDH-deficient GIST cells leads to a loss of over a thousand potential CTCF binding sites[107]. Importantly, the expression of genes adjacent to the affected CTCF binding sites changes significantly, indicating that SDHdeficient cells similarly acquire promiscuous enhancer-promoter interactions and an altered genome topology. Expression ofKIT, one the most frequently mutated RTKs in most GISTs but not WT-GISTs[1], is significantly upregulated by the altered CTCF binding profile[107]. This discovery raised the interesting possibility that activation of KIT and downstream signaling pathways are an underlying cause of both RTK-mutant and WT GISTs.
In addition to affecting KIT, disruption of CTCF binding increased the expression ofFGF3andFGF4, both of which are known to drive human cancer[42]. FGF3 and FGF4 are ligands activating the tyrosine kinase receptor FGFR, which plays a known role in the pathogenesis of GISTs[108,109]. Expression analysis revealed that KIT and FGFR are the most abundantly expressed RTKs in SDH-deficient GISTs. CTCF binding to the recognition motifs adjacent toFGF3andFGF4genes is disrupted in SDH-deficient GISTs, which leads to an increase inFGF3andFGF4expression by several magnitudes[110]. The upregulation ofFGF3andFGF4likely plays a redundant role with the gain-of-function mutation ofKITbecause both activate the MAPK pathway[108,109]. KIT inhibition by imatinib showed no efficacy in treating WT GISTs[6-8]. Interestingly, combinatorial treatment with the FDA-approved KIT inhibitor sunitinib and the FGFR inhibitor BGJ-398 effectively reduced SDH-deficient GISTs in a patient-derived xenograft (PDX) mouse model[107]. More intriguingly, while treatment with sunitinib alone had only a marginal effect, which is consistent with the previous observation that WT-GISTs are refractory to KIT inhibitors[7], treatment with BGJ-398 could completely suppress tumor growth in this model[107].
Oncometabolites regulate histone methylation:Jumonji domain-containing lysine demethylases (KDMs) belong to the α-KG-dependent dioxygenase family and can be inhibited by succinate, fumarate, and D-2HG[77-79]. Loss-of-function or reduced expression of Jumonji domain-containing KDMs has been correlated with several human cancers, such as renal cell carcinoma, GBM, and multiple myeloma[111-113]. These KDMs remove the methyl group on lysine in histone tails, which can either activate or repress transcription depending on the specifically modified lysine residues. For example, these oncometabolites inhibit the activities of both KDM4A and KDM7A, which remove methylation on histone H3K36 and H3K9, respectively[77,78]. Methylation of histone H3K36 often occurs in the gene body region and is known to antagonize silencing and safeguard transcription fidelity[114], while methylation of H3K9 has been associated with transcriptional silencing and heterochromatin formation[94]. Therefore, the impact of oncometabolite accumulation on histone tail modification is complex. Nonetheless, dysregulation of histone methylation has been associated with poor prognosis of human malignancies such as prostate cancer, lung cancer, breast cancer, colon adenocarcinoma, and gastric adenocarcinoma[65,115-117]. Experimental inactivation ofSDHandFHcauses increased H3K9 methylation in paraganglioma, pheochromocytoma and smooth muscle tumors[118]. In LGGs,IDHmutations suppress the histone H3K9 demethylase KDM4C[119]. LGG cells exhibit increased H3K9 methylation and downregulation of the glial differentiation transcriptional program, suggesting thatIDHmutation-induced D-2HG accumulation leads to a differentiation blockage of LGG cells[119,120]. However, the specific target genes affected by oncometabolites that are critical in tumorigenesis remain largely unknown. Recently, inhibition of H3K9 demethylases inIDHmutation-bearing LGGs and AMLs was found to reduce the expression of the ataxia telangiectasia mutated (ATM) gene[121], which encodes a key regulator involved in the DNA damage response (DDR) pathway. Consequently, AML cells withIDHmutations are defective in DDR responses and accumulate unrepaired DNA damage[121]. SDH-deficient hereditary paraganglioma and pheochromocytoma are also known to have reduced capacities in the DDR pathway[122]. However, whether SDH deficiency similarly leads to reduced expression ofATMand whether an impaired DDR contributes to the tumorigenesis of GISTs are still unknown, and these questions represent directions for future investigation.
ONCOMETABOLITES AND THE HYPOXIA RESPONSE PATHWAY
Activation of the hypoxia response pathway by overexpression of hypoxia-inducible factor 1α (HIF1α) plays an important role in the progression of SDH-deficient tumors[11,45,123,124]. Under normoxic conditions, HIF1α turns over rapidly by proteasomal degradation that is dependent on the ubiquitin E3 ligase VHL[125]. The VHL recognition of HIF1α requires hydroxylation on proline residues on HIF1α, which is mediated by prolyl hydroxylases (PHDs)[125]. Under hypoxic conditions, HIF1α degradation is blocked because the enzymatic activities of PHDs are suppressed by low levels of oxygen. HIF1α then translocates into the nucleus, forms the HIF1 heterodimer complex with HIF1β, and activates the hypoxia response pathway[125]. Transcriptional targets of HIF1 control several aspects of tumor biology, including angiogenesis, proliferation and survival, glucose metabolism, invasion, and metastasis[124-130]. Because PHDs belong to the α-KG-dependent dioxygenase family, oncometabolites can effectively activate the hypoxia response pathway by competitively inhibiting PHDs under normoxic conditions. Forced expression of LGG-inducingIDHmutations elevated levels of HIF1α and its transcriptional targets in cells, which is consistent with the observation that the HIF1α pathway is more active inIDH-mutant glioma patient samples[131]. Similarly, HIF1α and its transcriptional targets, such asVEGFandIGF, are more abundantly expressed in SDH-deficient GISTs than in RTK-mutant GISTs[45,132,133]. These observations suggest that the hypoxia response pathway is more active in SDHdeficient GISTs than in GISTs with normal SDH activity, which is likely caused by succinate-mediated PHD inhibition.
OTHER PATHWAYS REGULATED BY ONCOMETABOLITES
Oncometabolites and RNA methylation
RNA methylation, particularly the N6-methyladenine (m6A) modification in mRNA, has emerged as an important regulatory mechanism of RNA metabolism in recent years[134]. Genome-wide profiling studies have demonstrated that mRNA produced by approximately 15000 human genes has m6A modifications, which affect the splicing, stability, and translation efficiency of the modified mRNA molecules[135,136]. The m6A modification is deposited on mRNA molecules by the RNA methyltransferase writer enzyme complex METTL3-METTL14, removed by the eraser enzymes FTO and ALKBH5, and decoded by reader proteins such as YTHDF1 and YTHDF2[134]. Dysregulation of m6A modification has been suggested to drive human cancers[137]. Among the genes involved in m6A modification are the eraser enzymes FTO and ALKBH5, which belong to the α-KG-dependent dioxygenase family[138]. It has been demonstrated that the enzymatic activity of FTO is inhibited inIDHmutation-bearing AML cells, which correlates with a significant increase in m6A levels[139]. The inhibition of FTO in AML cells seems to have a positive effect on all-trans retinoic acid (ATRA)-induced leukemic cell differentiation[139]. Given the similar inhibitory roles ofIDHmutations and SDH deficiency, it is highly expected that SDH-deficient GISTs will have similarly elevated m6A levels. However, how such changes may contribute to tumorigenesis and the treatment of GISTs is awaiting future investigation.
Oncometabolites and repair of DNA alkylation damage
The alkB homolog (ALKBH) family enzymes are α-KG-dependent dioxygenases involved in repairing DNA damage caused by alkylating agents[140,141]. InIDH-mutant LGG cells, the activities of ALKBH enzymes are suppressed, which makes these cells sensitive to alkylation damage[142]. Consistently, combinatorial treatment of the DNA alkylating agents procarbazine and lomustine with the microtubule polymerization inhibitor vincristine is beneficial in some glioma patients[143,144]. In contrast, experimental restoration of the enzymatic activities by forced expression ofALKBHfamily members inIDH-mutant cells blunts the responses to alkylating agents[142]. However, the activities of ALKBH enzymes in SDH-deficient GISTs and the benefit of their inhibition in treating these tumors have not yet been investigated.
Protein succination
Oncometabolites may also function in mechanisms independent of α-KG-dependent dioxygenases. Because of the structural similarity, D-2HG produced byIDHmutations competitively inhibits SDH[145]. Inhibition or genetic inactivation of SDH results in the accumulation of succinate, which increases levels of succinyl-CoA, a metabolite immediately upstream of succinate and fumarate in the TCA cycle (Figure 1)[145]. Succinyl-CoA is the substrate for lysine succinylation, a common posttranslational modification that affects cellular stress responses[146]. In tumor cells withIDHorSDHmutations, proteins are preferentially hypersuccinylated in the mitochondria, which induces mitochondrial depolarization and cancerous metabolism[145]. Hypersuccinylation confers tumor cells with apoptosis resistanceviadeposition of the prosurvival factor BCL-2 onto the mitochondrial membrane[145]. Notably, inhibition of hypersuccinylation by overexpression of SIRT5, the enzyme that reverses succinylation, slowsIDHmutation-bearing tumor growth in a mouse xenograft
model[145]. The best-understood downstream target regulated by protein succinylation is the KEAP1-NRF2 pathway[147,148]. NRF2 regulates the transcription of genes involved in the antioxidant response. Binding of KEAP1 leads to degradation of NRF2, while the succinylation of KEAP1 prevents KEAP1 binding, therefore activating the NFR2-mediated transcriptional program[147]. It has been estimated that approximately onethird of all nucleosomes have succinylated lysine residues in their histone tails. Histone succinylation has been correlated with active gene transcription[149]. However, despite the broad impact on gene expression, how histone and nonhistone protein succinylation affects tumorigenesis remains largely unexplored.
TREATMENT OF SDH-DEFICIENT GISTS
GISTs with RTK mutations are resistant to standard chemotherapy but show responsiveness to the first-line KIT inhibitor imatinib[150]. Because of the lack of RTK mutations, WT GISTs, including all SDH-deficient GISTs, are resistant to the firstgeneration RTK inhibitor imatinib[6-8]. However, second-generation RTK inhibitors, such as sunitinib, have shown only moderate effectiveness in SDH-deficient GISTs[151]. Patients with SDH-deficient GISTs are recommended to undergo complete tumor resection[9,10]. As suggested by studies in preclinical animal models[107], SDH-deficient GISTs may be more responsive to combinatorial treatment with the KIT inhibitor sunitinib and an FGFR inhibitor than to monotherapy. This serves as a great example of how research on the basic biological mechanism may shed light on the potential treatment of human diseases.
CONCLUSION
Cancer is a complex disease that involves genetic mutations in multiple genes. The connection between altered cellular metabolism and tumorigenesis has gained increased attention in recent years. The discovery that mutations in metabolic enzymes such as IDH, SDH, or FH mutations drive tumorigenesis indicates that metabolic reprogramming likely plays an underappreciated role in human cancer. Oncometabolites such as D-2HG, succinate, and fumarate competitively inhibit α-KGdependent dioxygenases, which has a profound impact on epigenetic status, hypoxia response, RNA metabolism, and DNA repair capacity (Figure 2). We are only beginning to understand how these metabolic enzyme mutations and oncometabolites promote cancerous changes at the molecular and cellular levels. Further elucidation of the underlying mechanisms will lead to discoveries of novel approaches to treat SDHdeficient GISTs and other tumors with similar mutations.
ACKNOWLEDGEMENTS
We sincerely thank Drs. Rui Ji, Zhao-Feng Chen, Xi Gou, and Jun Wang for their insightful discussion.
杂志排行

World Journal of Gastroenterology的其它文章
- Acupuncture improved lipid metabolism by regulating intestinal absorption in mice
- Experimental model standardizing polyvinyl alcohol hydrogel to simulate endoscopic ultrasound and endoscopic ultrasound-elastography
- Efficacy of pancreatoscopy for pancreatic duct stones: A systematic review and meta-analysis
- Construction of a convolutional neural network classifier developed by computed tomography images for pancreatic cancer diagnosis
- Mixed epithelial endocrine neoplasms of the colon and rectum – An evolution over time: A systematic review
- Transjugular intrahepatic portosystemic shunt for Budd-Chiari syndrome: A comprehensive review