超高效液相色谱-静电场轨道阱高分辨质谱法测定法莫替丁及其制剂中的痕量N-亚硝基二甲胺
2020-09-29郭常川杨书娟王维剑文松松徐玉文
郭常川,杨书娟,刘 琦,王维剑,文松松,牛 冲,徐玉文
(山东省食品药品检验研究院 国家药品监督管理局仿制药研究与评价重点实验室 山东省仿制药一致性评价工程技术研究中心,山东 济南 250101)
N-亚硝基二甲胺(N-nitrosodimethylamine,NDMA)又名N-二甲基亚硝胺,具有很强的致癌、致畸和致突变毒性,在ICH M7指南中明确说明该化合物具有强致癌性[1]。根据世界卫生组织(WHO)公布的致癌物清单,NDMA属于2A类致癌物质[2]。自2018年以来,国内外多家药企涉及了缬沙坦、雷尼替丁等药品中NDMA污染事件,甚至启动了全球召回程序。2020年4月1日,美国食品药品管理局(FDA)发布公告要求制药商立即撤回所有雷尼替丁药物,涉及处方药和非处方药,这是全球针对雷尼替丁NDMA污染调查的最新重磅举措。法莫替丁化学结构中具有与雷尼替丁类似的遗传毒性、致癌性警示基团,因此很有必要对法莫替丁中的NDMA含量进行调查。
2020年5月8日,中国国家药品监督管理局(NMPA)发布了《化学药物中亚硝胺类杂质研究技术指导原则(试行)》[3],分析了药品中亚硝胺类杂质引入的原因,并规定了亚硝胺类杂质的限度控制。目前,NDMA 的检测方法主要为高效液相色谱法(HPLC)[4]、HPLC-化学发光检测法[5]、表面增强拉曼散射法(SERS)[6]、分子印迹聚合物电化学检测法[7]、分子印迹聚合物液质联用检测法[8]、超临界流体色谱法[9]、二维离子色谱法[10]、液相色谱-质谱联用法(LC-MS)[11-12]、气相色谱-热能分析法[13]、气相色谱-质谱联用法(GC-MS)[13-15]等。但这些方法主要应用于食品(饮用水、水产品、肉制品等)、化妆品和烟草行业,而药品中NDMA 检测的研究极少,目前仅有测定缬沙坦、厄贝沙坦中NDMA的文献报道[16-18],但对雷尼替丁、法莫替丁中NDMA 的检测至今未见报道。然而,由于原料药不同,其理化性质不同导致提取方法、色谱保留行为均有差异,因此测定其它药物中NDMA的文献方法不能直接应用于法莫替丁中NDMA的测定,亟需重新开发方法。静电场轨道阱(Orbitrap)高分辨质谱技术由于其高灵敏度、高质量精度的优势,在药物杂质分析领域应用广泛[19]。本研究首次建立了用于测定法莫替丁及其制剂中NDMA含量的超高效液相色谱-静电场轨道阱高分辨质谱法(UHPLC-Orbitrap HRMS),样品前处理简便快速,灵敏度高、专属性强,已用于法莫替丁及其制剂中NDMA的测定。
1 实验部分
1.1 仪器、试剂与材料
Thermo Q Exactive PlusTM超高效液相色谱-质谱联用系统包括Ultimate 3000液相泵、自动进样器、柱温箱以及Orbitrap高分辨质谱部分(美国Thermo Fisher Scientific Inc.);XCalibur 4.0软件(美国Thermo Fisher Scientific Inc.)用于质谱仪控制和数据处理;Mettler XS205型电子天平(瑞士Mettler-Toledo LLC);Heraeus Multifuge X1R型高速冷冻离心机(美国Thermo Fisher Scientific Inc.);KQ-500 DE型温控超声仪(昆山市超声仪器有限公司);THZ-82型水浴恒温振荡器(常州金坛精达仪器制造有限公司);2 mL一次性使用无菌注射器(山东新华安得医疗用品有限公司);13 mm、0.22 μm 微孔滤头(上海安谱实验科技股份有限公司)。
NDMA对照品(含量为99.5%,TCI公司);甲醇、乙腈、甲酸均为HPLC级(美国Thermo Fisher Scientific Inc.);实验用纯水(18.2 MΩ·cm)由Millipore Milli-Q Advantage A10超纯水系统(美国Merckmillipore Inc.)制得。11批法莫替丁原料、36批法莫替丁钙镁咀嚼片(每片含法莫替丁10 mg)及辅料均为药企委托检验提供。
1.2 对照品及供试品溶液的制备
1.2.1 对照品溶液取NDMA对照品适量,精密称定,置于50 mL容量瓶中,加甲醇溶解并稀释至刻度,得1.00 mg/mL的NDMA对照品储备液;精密移取上述储备液1.0 mL于100 mL容量瓶中,加甲醇定容至刻度,摇匀得10.00 μg/mL的对照品溶液;同法以甲醇逐步稀释,得1.00、2.00、5.00、10.00、50.00、100.00 ng/mL的系列质量浓度对照品溶液。
1.2.2 供试品溶液法莫替丁供试品溶液(1.00 mg/mL):取供试品约10 mg,精密称定,置于50 mL离心管中,加入10 mL甲醇,涡旋混匀1 min使其完全溶解,即得。
法莫替丁钙镁咀嚼片供试品溶液(1.00 mg/mL):取本品5片,精密称定,研细,称取细粉适量(约相当于法莫替丁10 mg)至50 mL离心管中,加入10 mL甲醇,涡旋混匀1 min,再以350 r/min振摇40 min,于10 000 r/min、4 ℃条件下离心10 min,上清液过0.22 μm微孔滤膜,取续滤液即得。
1.3 色谱条件
色谱柱为ACE EXCEL 3 C18-AR(150 mm × 4.6 mm,3 μm),流速:0.50 mL/min;柱温:30 ℃;自动进样器温度:4 ℃;进样量:5 μL。流动相:A为0.1%甲酸水溶液,B为0.1%甲酸乙腈。梯度洗脱程序:0~1.0 min,5%B;1.0~3.0 min,5%~20%B;3.0~7.0 min,20%~100%B;7.0~9.0 min,100%B;9.0~9.1 min,100%~5%B;9.1~14.0 min,5%B。六通阀切换设置:保留时间5.00~7.00 min的流动相进入质谱,其余时间的流动相进入废液。按外标法以峰面积计算供试品中NDMA含量。
1.4 质谱条件
电喷雾离子源(ESI),采用正离子平行反应监测(PRM)扫描模式,扫描范围:m/z50~95,扫描m/z:75.055 6,隔离窗口:m/z1.5,碰撞能量(NCE):30。鞘气流速:55 arb(arbitrary units),辅助气流速:15 arb,喷雾电压:3.5 kV,离子传输管温度:350 ℃,辅助气加热温度:400 ℃。
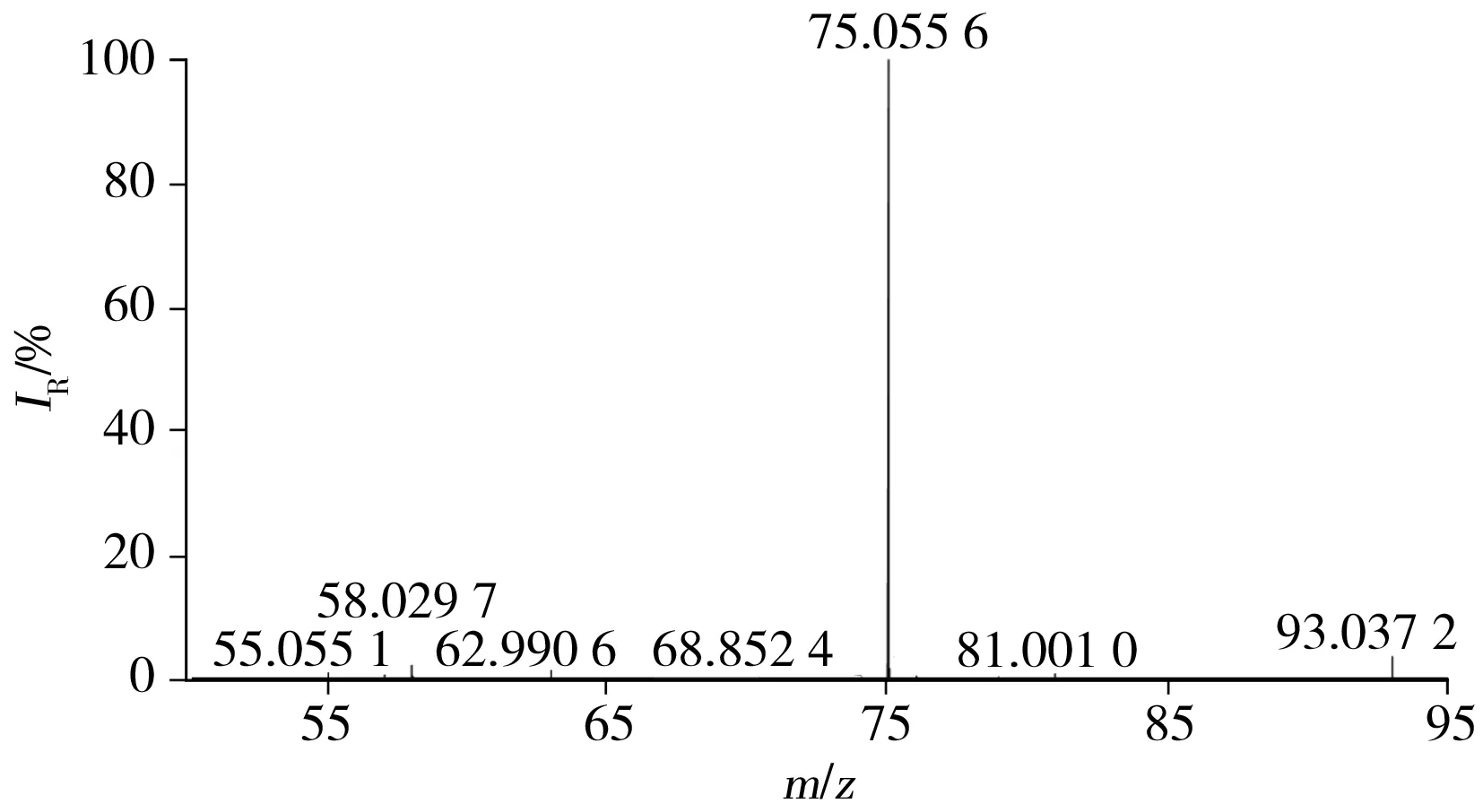
图1 NDMA的PRM模式高分辨质谱图Fig.1 High resolution mass spectrum of NDMA in PRM mode
2 结果与讨论
2.1 实验条件的考察
2.1.1 质谱条件的优化《化学药物中亚硝胺类杂质研究技术指导原则(试行)》[3]规定NDMA的可接受摄入限度为96 ng/d,而法莫替丁钙镁咀嚼片的最大摄入量为20 mg/d,二者的比值4.80×10-6(即4.8 ppm)即为法莫替丁及法莫替丁钙镁咀嚼片中NDMA的允许限度。根据供试品溶液中法莫替丁的质量浓度为1.00 mg/mL,NDMA的定量下限须低于2.40 ng/mL(即NDMA限度浓度4.8 ng/mL的50%),对仪器灵敏度性能要求很高。但NDMA是低质量数化合物,其母离子m/z仅为75.055 6,质谱检测时基线噪音较大,影响色谱峰信噪比,导致灵敏度较低。为此,考察了全扫描(Full MS)、目标物选择性离子检测(Targetd SIM)、全扫描-数据依赖的二级扫描(Full MS/dd MS2)、平行反应监测(PRM)等不同的质谱扫描模式,发现PRM模式下NDMA的基线噪音最低、信噪比最高,其信噪比高于其他扫描模式10倍以上。应用XCalibur 4.0软件选择最佳碰撞能量、提取离子,并对离子源气流速、温度、电压进行优化,最终实现了NDMA检测的灵敏度要求。最佳质谱条件如“1.4”所示,NDMA的高分辨质谱图如图1所示。
2.1.2 色谱条件的优化供试品溶液中含有高质量浓度的法莫替丁(1.00 mg/mL)和痕量的NDMA(<100.00 ng/mL),如果高浓度法莫替丁进入质谱系统,会污染离子源并产生严重的基质效应,影响痕量NDMA的准确测定。因此,应使NDMA和法莫替丁达到足够的色谱分离度,通过六通阀将法莫替丁色谱峰切换到废液,避免高浓度法莫替丁进入质谱。考察了ACE EXCEL 3 C18-AR(100 mm×4.6 mm,3 μm)色谱柱的分离效果,发现NDMA和法莫替丁的色谱分离较差,二者保留时间仅相差1.3 min,对阀切换造成了困难。改用ACE EXCEL 3 C18-AR(150 mm×4.6 mm,3 μm)色谱柱,NDMA的保留时间约为6.15 min,与主成分法莫替丁的保留时间差值增至2.4 min,实现了较好的色谱分离,通过六通阀仅将保留时间5.00~7.00 min的流动相进入质谱,有效避免了高浓度法莫替丁的基质干扰,保护了离子源,使NDMA的测定更加准确可靠。优化的色谱条件如“1.3”所示。
2.1.3 样品预处理方法的优化NDMA易溶于水、醇,但法莫替丁在冰醋酸中易溶,在甲醇中微溶,在水中几乎不溶。本实验分别以冰醋酸和甲醇作为溶剂配制1.00 mg/mL的法莫替丁和5.00 ng/mL NDMA混合溶液,平行测试6次(n=6)。结果显示,冰醋酸中NDMA的平均色谱峰面积仅为在甲醇中峰面积的34.2%。原因可能为暴露在酸中的NDMA在前处理过程中不稳定所致。因此,本实验选用甲醇作为提取溶剂,并控制称样量使主成分法莫替丁的质量浓度为1.00 mg/mL(浓度过高则法莫替丁过饱和)。此外,还考察了振摇时间对NDMA提取的影响,发现振摇20、40、60、90 min时,阴性样品加标溶液中5.00 ng/mL NDMA的色谱峰面积分别为421 078、497 403、518 269、513 247,提取时间为40、60、90 min时的NDMA峰面积无显著差异,故选择振摇时间为40 min。本实验前处理简单,提取效果好。
2.2 方法学验证
2.2.1 专属性将溶剂(甲醇)、处理后的辅料溶液、NDMA对照品溶液、法莫替丁溶液、加标回收溶液分别进样测定,NDMA的出峰时间为6.16 min,相同保留时间处的溶剂及辅料溶液均无干扰峰。辅料溶液、NDMA对照品溶液和加标回收溶液的提取离子色谱图见图2。法莫替丁的保留时间为8.57 min,对NDMA色谱峰无干扰。以上结果表明本方法的专属性良好。
图2 提取离子色谱图Fig.2 Extracted ion chromatogramsA:excipients;B:NDMA standard solution;C:sample of recovery test
2.2.2 基质效应分别制备低(2.50 ng/mL)、中(5.00 ng/mL)、高(7.50 ng/mL)3个水平的空白基质加标溶液和对照品标准溶液,分别进样测定,计算两种溶液中NDMA响应值的百分比,即为基质效应。结果表明,NDMA在低、中、高3个不同浓度下的基质效应为91.4%~102.7%,表明基质不影响NDMA的质谱测定。
2.2.3 线性范围将“1.2.1”配制的系列浓度对照品溶液进样测定。以所得峰面积为纵坐标(y),NDMA标准溶液的质量浓度为横坐标(x,ng/mL),加权重1/x2拟合线性校正曲线,建立回归方程,并计算相关系数(r)。结果表明,NDMA在1.00~100.00 ng/mL质量浓度范围内线性关系良好,回归方程为y=98 665x-19 485,相关系数r=0.999 7。
2.2.4 检出限与定量下限精密移取1.00 ng/mL的NDMA标准溶液,以甲醇为溶剂逐级稀释,采用本方法进行测定,按信噪比S/N≥3计算检出限,S/N≥10计算定量下限。结果表明方法的检出限为0.20 ng/mL,定量下限为1.00 ng/mL。
2.2.5 回收率与相对标准偏差考察低、中、高3个浓度的NDMA在法莫替丁及其制剂中的回收率和相对标准偏差(RSD)。取已知未检出NDMA的阴性样品约1次服用量(以法莫替丁计10 mg),法莫替丁原料药(API)及制剂均精密称取9份(低、中、高水平各3份),分别精密加入适量的NDMA对照品溶液,后续操作按照“1.2.2”方法处理,进样测定,按外标法计算NDMA含量。由表1可知,3个加标浓度下NDMA在法莫替丁及制剂中的平均回收率为98.5%~108%,RSD为2.3%~6.7%,符合药典规定,表明NDMA的回收率和重复性良好。
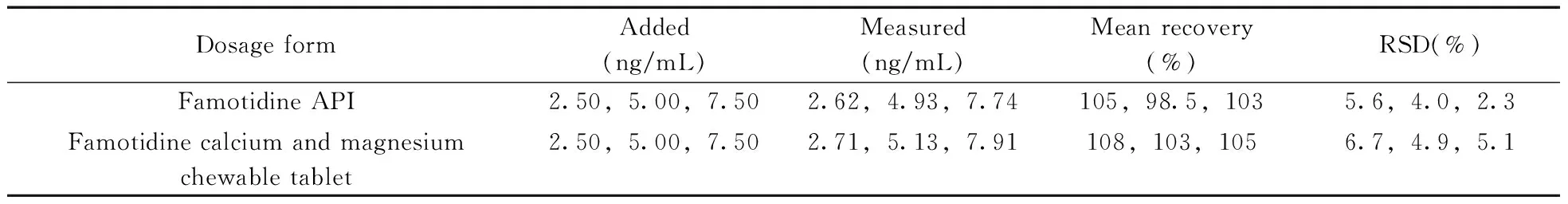
表1 NDMA在法莫替丁及其制剂中的回收率及相对标准偏差(n=3)Table 1 Recoveries and RSDs of NDMA in famotidine API and preparations(n=3)
2.2.6 稳定性将2.00 ng/mL的NDMA标准溶液保存于进样瓶中,在自动进样器中按照实验条件放置0、4、8、12、24、48 h后进样测定,记录色谱图和峰面积。结果显示,NDMA峰面积的RSD为6.9%,说明该条件下NDMA在48 h内稳定性良好。
2.2.7 耐用性采用2.00 ng/mL的NDMA标准溶液考察了方法的耐用性。将超高效液相色谱-质谱联用仪的柱温分别调整为27、33 ℃,流动相流速分别调整为0.45、0.55 mL/min时,NDMA的色谱图峰形无变化,保留时间和峰面积发生微小改变,表明测定条件轻微变动不影响NDMA的测定,方法耐用性良好。
2.3 样品测定
将47批法莫替丁及制剂样品按照“1.2.2”方法制备供试品溶液,按照“1.3” 和“1.4”条件进样测定,记录色谱图和峰面积,外标法计算NDMA含量。按照法莫替丁中NDMA的限度要求4.8×10-6(即4.8 ppm),在1批法莫替丁原料和2批法莫替丁制剂中检出了超限的NDMA,测得其质量浓度为6.77~9.73 ng/mL,含量为6.62×10-6~9.57×10-6(即6.62~9.57 ppm)。阳性样品的测定结果如表2所示。
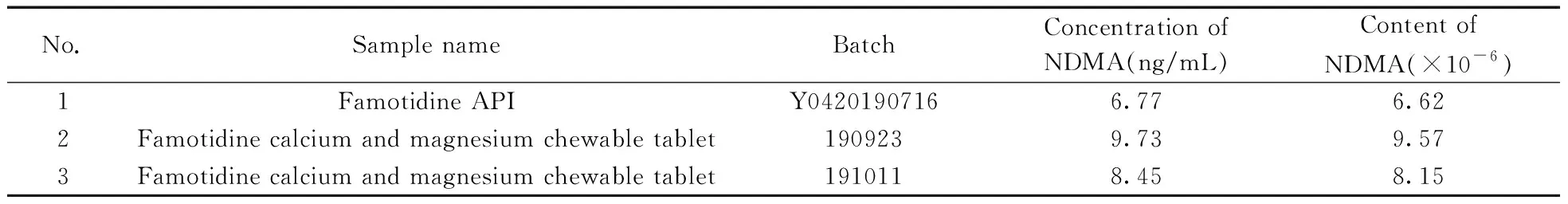
表2 阳性样品的测定结果Table 2 Determination results of positive samples
3 结 论
本研究建立了法莫替丁中NDMA的UHPLC-Orbitrap HRMS法,进行了方法学验证并将方法用于法莫替丁及其制剂中NDMA的测定。NDMA的线性范围为1.00~100.00 ng/mL,检出限和定量下限分别为0.20 ng/mL和1.00 ng/mL,低、中、高3个加标浓度下的平均回收率为98.5%~108%,RSD为2.3%~6.7%。将该法用于47批供试品中NDMA的测定,在1批法莫替丁原料和2批法莫替丁制剂中检出了超限的NDMA。本方法灵敏度高、专属性好、回收率高、线性范围宽,操作简便、快速,已用于法莫替丁及其制剂中NDMA的检验,填补了该研究技术空白。本研究有助于医药企业进行生产工艺控制,并为药监部门的监管提供有力的技术支持。
