铂基金属间化合物纳米晶的最新进展:可控合成与电催化应用
2020-09-29杨天怡崔铖戎宏盼张加涛王定胜
杨天怡,崔铖,戎宏盼,*,张加涛,王定胜
1北京理工大学材料学院,结构可控先进功能材料与绿色应用北京市重点实验室,北京 100081
2清华大学化学系,北京 100084
1 引言
可持续绿色能源的开发利用是解决当前气候和环境问题的关键。风能、太阳能等可持续绿色能源目前存在波动问题,需要通过能源转换存储来解决波动问题以适应人类需求1–7。通过电化学将风能、太阳能等转化为化学能存储是一种有效的解决方式。通常,此过程涉及的反应有阴极上发生的析氢反应(HER)和阳极上发生的析氧反应(OER)。
燃料电池具有高比能量、低环境污染等特点,近年来受到了广泛关注。其能量来源,可以是氢气、甲酸、甲醇和乙醇等。这些含氢类分子在阳极发生氧化反应时,阴极同时发生氧还原反应(ORR)。为了使这些能量转化过程达到最大化的效率和最小化的能量损失,科学家致力于研究各类反应机理并设计优化催化剂。对于上述大多数反应,铂(Pt)元素表现优异,但是常常需要使用高负载量的Pt催化剂才能使燃料电池达到较高的功率密度8。例如,燃料电池汽车中的Pt负载量通常高达0.3–0.4 mg.cm−2,丰田燃料电池汽车Mirai使用的PtCo/C催化剂,阴极和阳极的铂负载量分别为0.315和0.05 mg.cm−2,远高于美国能源部2020年的技术指标(表1)9,10。一方面,Pt价格昂贵,受到成本和资源制约;另一方面,高载量的Pt在电催化条件下循环多次后容易聚集,降低催化剂的比表面积导致比活性下降,吸附的CO等物种更会使催化剂失活11–14,阻碍了其广泛的商业化应用。近年来,将非贵金属与Pt合金化(如PtNi,PtFe和PtCo等合金)是有效降低催化剂成本,提高催化剂性能的策略之一,然而这种合金常存在稳定性差等问题,这是因为非贵金属在苛刻的电催化环境下极易被腐蚀造成去合金化现象。
不同于传统合金催化剂中各种金属元素的随机分布,在金属间化合物结构中,二元或多元金属原子之间通过d轨道强相互作用形式键连在一起,并在特定晶格方向进行长程有序排列。这种有序结构使得金属间化合物在热力学上更为稳定15,从而在电催化反应中表现出比相应的合金材料更好的抗氧化,耐腐蚀和耐CO中毒的能力16。将金属间化合物制成高分散的纳米晶,由于其超大的比表面积和体积效应可以显著提升其活性位点密度,并满足燃料电池对催化剂均匀分布和小粒径尺寸的要求。因此,利用非Pt金属对Pt的电子效应和几何效应,将二者组合制备金属间化合物纳米材料是降低催化剂成本,提升催化剂稳定性和活性的重要方法11。大量研究表明Pt基金属间化合物在HER17,18,ORR19–23,氢氧化反应(HOR)24,25,甲酸氧化反应(FAOR)26,27,甲醇氧化反应(MOR)6,7,28–33和乙醇氧化反应(EtOR)32反应中均表现出优异的活性和稳定性。下面从铂基金属间化合物纳米材料的可控合成策略、在电催化还原反应、氧化反应中的构效关系方面进行了总结,系统化阐述了铂基金属间化合物纳米结构的最新研究进展。
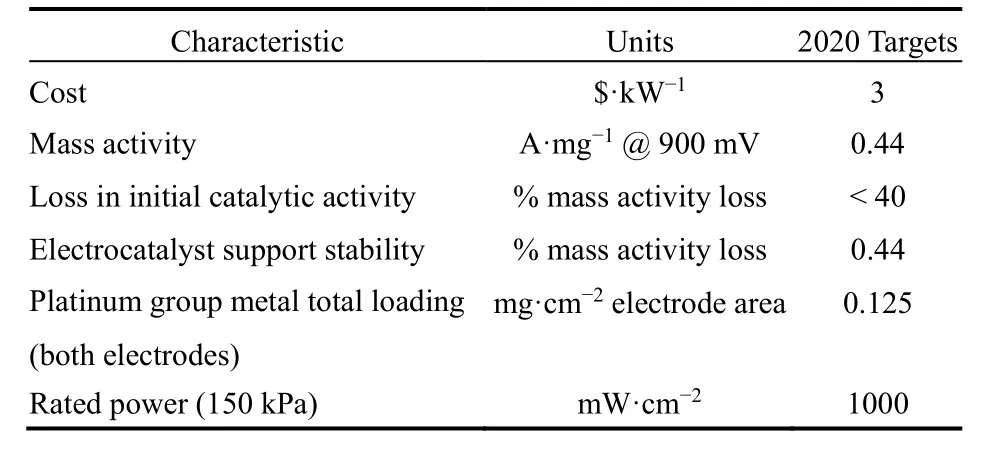
表1 美国能源部2020年质子交换膜燃料电池的技术目标9Table 1 DOE technical targets for polymer electrolyte membrane fuel cell components 9.
2 铂基金属间化合物纳米晶的可控合成
由于金属间化合物相的高度有序性及固体内部高扩散势垒,形貌均一的小尺寸金属间化合物纳米晶的制备困难重重。目前,后过渡金属、典型的金属或半金属(Zn,Ga,Mn,In,Sn,Sb,Fe,Co,Pb和Bi)元素与Pt形成金属间化合物的研究较多,而Pt与前过渡金属(如Sc,Ti,B,Y,Zr,Nb,La,Hf和Ta)元素的金属间化合物却鲜有报导。这是由于前过渡金属前驱体的还原电势非常负(通常< −1.5 VvsNHE (一般氢电极)),例如合成Pt3Ti或Pt3Nb需要900–1000 °C氢气气氛或液相体系中利用三乙基硼氢化锂等强还原剂还原34,35。而Sc3+和镧系离子的还原电势更负(通常< −2.0 VvsNHE),基本无法通过化学试剂还原。虽然Pt基前过渡金属化合物可以通过高温冶金制备合成,但暴露空气中后,其中包含的前过渡金属成分仍然极易被氧化导致金属间化合物结构破坏。因此本文主要对Pt和后过渡金属组成的金属间化合物进行总结与讨论。
传统的大块Pt基金属间化合物通常是通过粉末冶金、电弧熔炼或微波冶金等技术生产的,这些技术通常都包括高温退火过程。对于这些块体材料,研究者们很难通过破碎或球磨等后处理方法获得性能优异的、纳米尺寸的金属间化合物催化剂。近年来胶体化学的快速发展为探索金属间纳米晶体的合成奠定了坚实的基础36。目前,合成纳米尺寸的铂基金属间化合物的方法主要包括三大类(表2),即:直接液相合成法,高温诱导结构转变法和化学气相沉积法。其中常用的方法是直接在液相体系中合成有序结构,或者先在液相体系下可控的合成特定尺寸和成分的合金纳米颗粒(纳米晶),然后在合适的温度下进行退火处理,以形成有序的金属间化合物结构37。
2.1 直接液相合成
直接液相合成是基于溶液的化学合成方法,在典型的液相合成过程中,需要选择合适的化学反应条件(溶剂,表面活性剂)和相应的金属前驱体,才能优先生成目标产物。由于其简单且易扩大生产的特点,直接液相合成法被广泛应用于铂基金属间化合物的合成。由于溶剂沸点的限制,液相合成反应只能在低于结构转变所需的温度下进行。因此,研究者们必须通过降低结构转变能垒才能成功地在液相中直接合成金属间化合物纳米晶。常见的降低结构转变能垒的策略有两种:1)通过加入卤素离子(Cl−,Br−,I−)或有机酸(抗坏血酸)等具有腐蚀作用的试剂以减慢晶体生长速度;2)通过晶种诱导生长法控制第二种金属在核结构中的扩散。
2.1.1 溶剂热合成法
溶剂热法是在水热合成法的基础上发展而来的。具体而言是将金属前驱体、表面活性剂和溶剂置于密闭容器中加热,溶剂受热后在有限空间中产生高压,随着体系温度和压强的升高,金属前驱体受热分解或被还原为原子,达到一定浓度时爆发性成核,在表面活性剂的作用下以一定形貌生长并从溶液中析出。Pt基金属间化合物利用溶剂热法合成过程中,可能涉及一种或两种以上提到的腐蚀试剂和晶种诱导的两种策略,并利用反应釜中的高压环境促进金属间化合物结构有序化。溶剂热法可以有效合成具有均一形状、大小和组成的,分散性良好的铂基金属间化合物纳米材料26,32。
2014年,Chen等人38以乙酰丙酮铂(Pt(acac)2)和乙酰丙酮锌(Zn(acac)2)为金属前驱体,聚乙烯吡咯烷酮(PVP)作为表面活性剂,N,N二甲基甲酰胺(DMF)作为溶剂,在反应釜中加热至180 °C并保持9 h,成功合成了Pt3Zn金属间化合物。由于Zn(Zn/Zn2+,0.76 VvsSHE (标准氢电极))和Pt(Pt/Pt2+,1.18 VvsSHE)的还原电势不同,为了使Pt和Zn同时还原,就需要选择合适的Pt金属前驱体以降低Pt的还原电势。Pt(acac)2通过Pt2+与乙酰丙酮配体的络合,保持了缓慢的游离Pt2+供应,从而降低了Pt2+的还原速率。如果把Pt(acac)2换成氯铂酸(H2PtCl6)或氯铂酸钾(K2PtCl6)则只能得到PtZn合金,这是因为与Pt(acac)2相比,H2PtCl6或K2PtCl6有更正的还原电势和更快的还原速率。2016年,Rong等人26以Pt(acac)2和氯化亚锡(SnCl2.H2O)为金属前驱体,向反应釜中加入PVP和DMF,并加热至180 °C,在Cl−离子和氧气的腐蚀作用帮助下,通过水热合成法在反应釜中制备出了表面富缺陷的Pt3Sn立方块纳米晶。研究表明,一个锡原子被还原的同时两个Cl−离子被释放到溶液中。Cl−离子浓度较低时刻蚀缓慢,但随着被还原的Sn原子数量的增加,Cl−离子的析出量增加,刻蚀速率也随之增加,缺陷增多。新生成的锡原子可以从富含Pt的晶种表面扩散到晶格中,从而形成PtSn固溶体,反应釜中高温高压的环境为有序结构的形成提供了能量。研究表明,通过控制前驱体浓度调控动力学过程,可产生不同表面缺陷浓度的Pt3Sn金属间化合物纳米晶。

表2 近年来Pt基金属间化合物的合成方法Table 2 Synthesis methods of Pt based intergeneric compounds in recent years.
2.1.2 多元醇法
多元醇法也被用于金属间化合物的合成,多元醇(乙二醇,丙二醇和丙三醇等)沸点高(200–350 °C),在其中起到溶剂,封端剂和还原剂等多重作用39。多元醇法方法较为简单,已经被用于合成了几种金属间化合物纳米晶40。
2018年,Yuan等人32报道了一锅合成法来制备具有特定形状和尺寸的金属核壳纳米粒子—由金属间化合物Pt1Bi1作为核和超薄铂壳组成的PtBi/Pt核-壳纳米结构六方片。以Pt(acac)2和新十二酸铋(C30H57BiO6)分别作为Pt和Bi的前驱体,PVP为表面活性剂,二甘醇(DEG)为溶剂,搅拌条件下将金属前驱体和PVP与DEG混合并加热到150 °C保持30 min确保反应完全。后续的高分辨透射电镜(HRTEM)和X射线衍射(XRD)表征证明了其核-壳结构。通过元素分析研究产物形成的过程发现,反应1 min时得到的产物主要是由Pt元素和一些随机分布的Bi元素组成,说明早期率先形成的是富Pt种子,此后,随着反应时间的延长,Bi元素的含量会明显增加(图1)。直接在多元醇溶液中添加载体可制备负载的金属间化合物纳米晶。例如,Chen等人41将H2PtCl6.6H2O和四氯化锡(SnCl4.5H2O)作为前驱体加入到乙二醇(EG)中,在200 °C下反应2 h后,加入载体—含Sb的SnO2(ATO),成功制备出了ATO负载的PtSn金属间化合物纳米晶,该方法取得成功的关键是精确地控制了反应温度。与之类似地,Bauer等人42在不使用PVP的情况下,通过加热K2PtCl6的EG或四甘醇(TEG)溶液,用多元醇法制备了Pt纳米晶,然后将无负载的Pt纳米晶与适当的金属盐溶液在一定条件下还原转化为PtSn,PtPb,PtBi和FePt3等单分散的金属间化合物纳米晶。这种方法的优点是不需要额外的高温退火过程,也能在不使用表面活性剂的情况下控制纳米晶的形貌,从而有效避免表面活性剂对催化反应的不利影响。
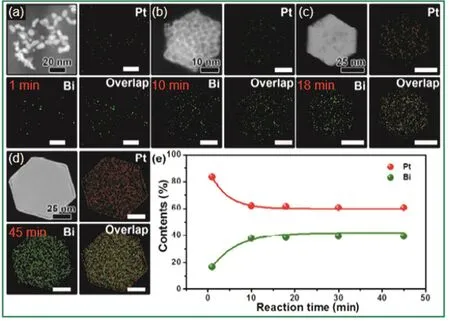
图1 150 °C下不同反应时间(a) 1 min,(b) 10 min,(c) 18 min,(d) 45 min产物中Pt和Bi元素的分布图32Fig. 1 Elemental mapping images showing the distribution of Pt and Bi after reacting at 150 °C for(a) 1 min, (b) 10 min, (c) 18 min, (d) 45 min 32.
2.1.3 油相高温法
油胺(OAm)和十八烯(ODE)混合溶剂由于沸点高,具有还原性,常被用于高温常压下制备油相Pt基金属间化合物纳米结构。2016年,Bu等人43使用OAm和ODE混合溶剂,160 °C下,以抗坏血酸(AA)为还原剂还原Pt(acac)2和乙酰丙酮铅(Pb(acac)2)。在此反应体系,Pb比Pt更易还原,Pt还原后沉积并扩散进入Pb晶格,最终形成Pt超薄壳层包覆PtPb金属间化合物核的六方片结构。2018年,Feng等人44在OAm和ODE混合溶剂体系300 °C还原Pt(acac)2和氯化镓(GaCl3)制备了约10 nm的Pt3Ga金属间化合物纳米立方体。将其负载于活性炭之后,用乙酸70 °C处理12 h除去了非晶态的GaOx和表面活性剂(OAm),并且腐蚀了纳米晶表面3–4原子层里的Ga,得到了Pt3Ga@Pt的核壳结构。此体系不仅可用于二元Pt基金属间化合物纳米晶的一步制备,还可以推广到制备三元Pt基金属间化合物纳米结构。例如,Luo等人45在OAm和ODE混合溶剂体系,在十六烷基三甲基溴化铵(CTAB)和AA的帮助下,还原Pt(acac)2,SnCl2和乙酸铋(Bi(act)3)一步制备了三元金属间化合物Pt45Sn25Bi30纳米六方片。
2.2 高温诱导结构转变
利用热退火处理合金纳米晶是目前获得金属间化合物纳米晶最常用的策略之一,高温过程可以改变纳米晶的形状结构,形成热力学稳定的有序多面体形状。Kim等人46发现合成的无序fcc(face centered cubic)结构的FePt合金纳米晶经750 °C热退火处理后相结构转化为有序fct (face centered tetragonal)结构的L10-FePt金属间化合物(L10结构如图216),并通过XRD表征证明了高温退火可以提高材料的有序度47。经过酸处理将纳米晶表面几个原子层的Fe除去形成L10-FePt@Pt核壳结构后,可以进一步稳定内部的Fe以提高催化剂的化学稳定性48。Wang等人49通过浸渍还原法制备的Pt3Co/C在700 °C高温H2/N2气流处理后,也转变为了金属间化合物相。
高温退火过程中经常会产生纳米晶聚集或烧结等问题,解决方法之一是将纳米晶负载在更稳定的载体上(如多孔碳、石墨烯、碳纳米管、多孔氧化物和碳氧化物)50,51。例如,超导电炭黑(Ketjenblack EC-300J carbon)支撑的CoPt纳米晶在650 °C热退火后结构由A1(结构见图2)转变为L1022。将Pt的前驱体滴加到二氧化硅负载的锰(Mn/SiO2)上,烘干后,通入氢气在550 °C下煅烧,获得了Pt3Mn金属间化合物13。
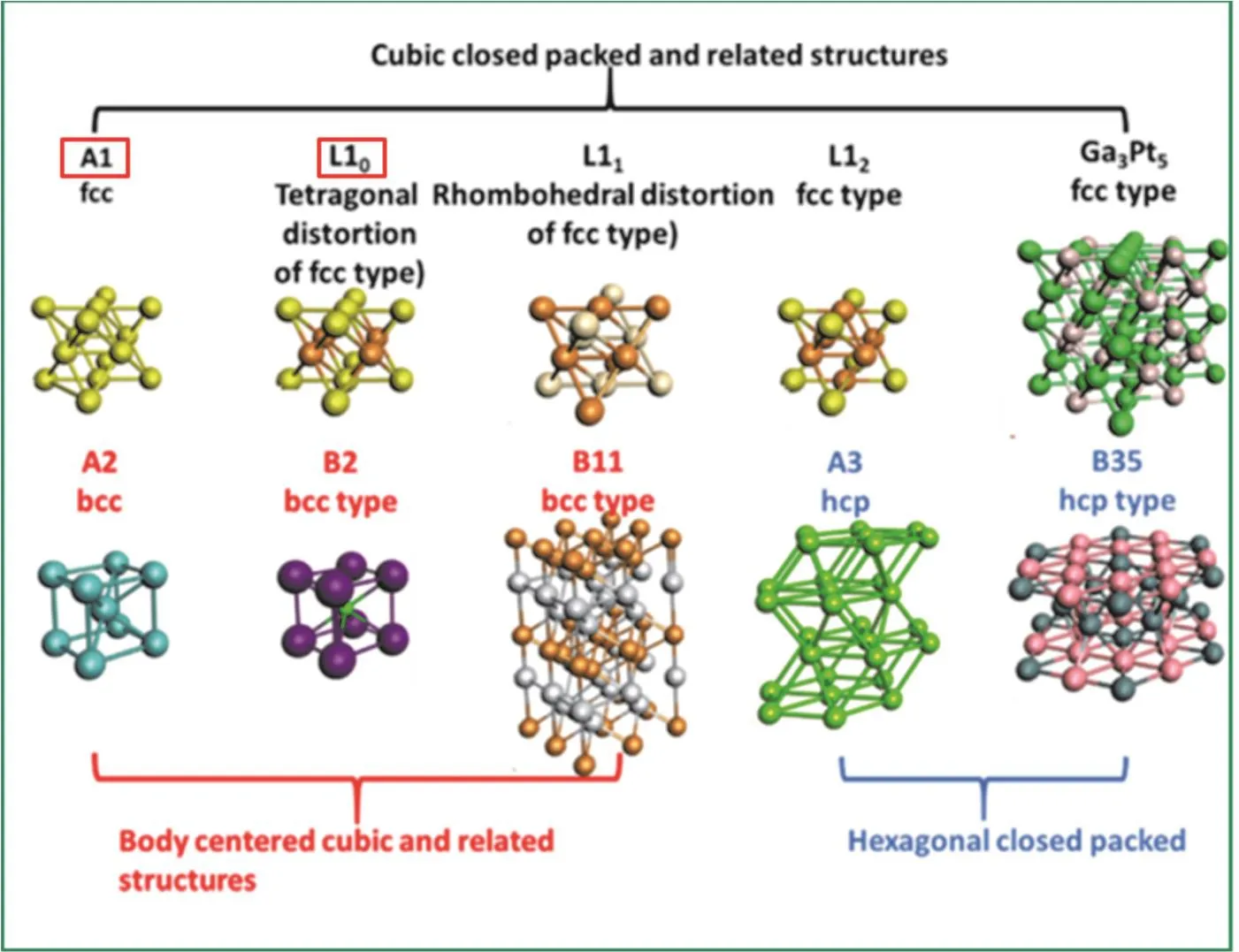
图2 常见合金结构及相应的金属间化合物结构16Fig. 2 Illustration of common alloy structures and their corresponding intermetallic structures 16.
采用在纳米晶表面添加包覆材料如介孔SiO2,MgO和聚合物等方法也能有效抑制退火过程中的纳米晶聚集和生长52。例如,介孔SiO2的包覆有效抑制了ZnPt合金纳米晶在高温环境转变为金属间化合物相过程中的聚集31,MgO的包覆在fcc合金相FePt纳米晶转化为fct的金属间化合物纳米晶过程中起到了重要的保护作用53–55。Han等人56首先在ZnO纳米棒上包覆一层聚多巴胺(PDA),加入K2PtCl6后通过调节pH将氢氧化铂(Pt(OH)4)沉积于PDA表面,再经800 °C氢气还原后形成2–6 nm的PtZn金属间化合物纳米晶,最后利用酸将内部ZnO腐蚀去除(图3)。在高温还原过程中,Pt与载体的强相互作用导致了PtZn金属间化合物的生成。
在高温退火过程中,包覆的材料如介孔SiO2,MgO和聚合物等可以有效地抑制纳米晶的聚集,但也降低了纳米晶中金属原子的扩散速率,增大了无序结构向有序结构的转化难度。为了减小包覆材料的不利影响,可以通过引入缺陷(空位或第三种元素)大幅降低纳米晶中金属原子迁移的势垒。Li等人57将哑铃状的Fe3O4-FePt纳米晶置于高温还原气氛下,将部分Fe3O4还原为Fe (形成氧空位),而氧空位的形成降低了金属原子扩散的势垒,从而促进了金属间化合物L10-FePt的生成。Zhang等人58发现fcc结构FePtAu合金中Au趋向纳米晶表面聚集,内部Au迁移留下空位促使金属间化合物L10-FePtAu生成势垒降低。利用相似的原理,当L10-FePt中的少量Fe被Cu替换后可得到金属间化合物L10-FeCuPt纳米晶59。
此外,利用载体与金属Pt的强相互作用,也可在高温情况下制备金属间化合物纳米晶。1997年,Bernal等人60观察到负载于CeO2上的Pt纳米晶在高温还原性气氛下转化为CePt5金属间化合物的现象。2019年,Li等人61也成功地将负载于二维过渡金属碳化物Ti3C2Tx(T代表表面终端物种如―O和―OH)表面的Pt纳米晶在高温下原位转化为Pt3Ti金属间化合物纳米晶。
2.3 化学气相沉积法
除了普遍使用的液相合成和高温诱导结构转变等方法,近几年化学气相沉积方法(CVD)也逐渐被应用于合成各种金属间化合物纳米晶(图4)。在Pt基金属间化合物纳米晶的CVD方法制备中,选择适当的温度,将容易挥发的第二金属前驱体在加热的基板上进行热分解或热反应,然后利用载气将第二种金属直接供给到事先制备并负载于惰性载体上的Pt纳米晶的表面,高温环境促进了第二种金属的还原沉积和合金化等过程的发生。因为纳米晶尺寸远远小于块状载体,较高的表面活化能导致第二种金属的还原沉积优先发生在Pt纳米晶表面,从而实现其选择性沉积。目前CVD法制备纳米合金的研究较多62,而合成金属间化合物的难度较大。
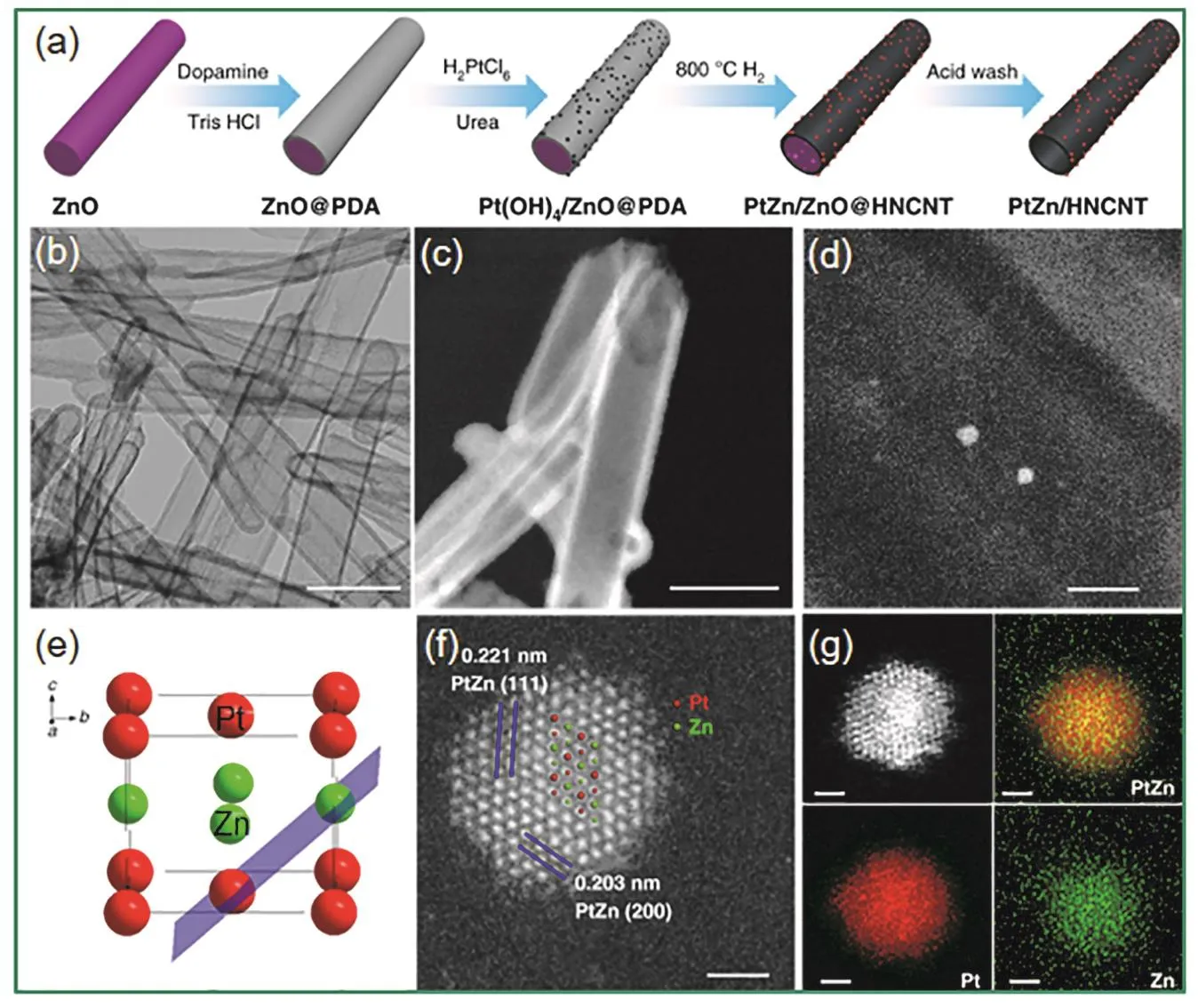
图3 中空氮掺杂碳纳米管负载的PtZn金属间化合物纳米晶的(a)合成示意图,(b) TEM图,(c)高角环形暗场扫描透射电子显微镜(HAADF-STEM)图,(d)球差校正的高角环形暗场扫描透射电子显微镜(AC-HAADF-STEM)图。(b),(c),(d)中的标尺分别为500,200,和10 nm。(e) PtZn金属间化合物的晶体结构,(f) AC-HAADF-STEM图像及(g)元素分布成像表征,(f),(g)中的标尺为1 nm 56Fig. 3 Synthetic scheme and characterization of the PtZn intermetallic nanoparticles. (a) Synthetic strategy of PtZn intermetallic nanoparticles supported on hollow nitrogen-doped carbon nanotubes (PtZn/HNCNT). (b–d) TEM(b) HAADF-STEM (c), and AC-HAADF-STEM (d) images of PtZn/HNCNT. Scale bar, 500 nm (b); 200 nm (c); 10 nm (d).(e) Crystal structure of PtZn intermetallic compound (IMC) (Pt: red; Zn: green). (f, g) AC-HAADF-STEM(f) image and elemental mappings (g) of a PtZn IMC nanoparticle. Scale bar, 1 nm 56.
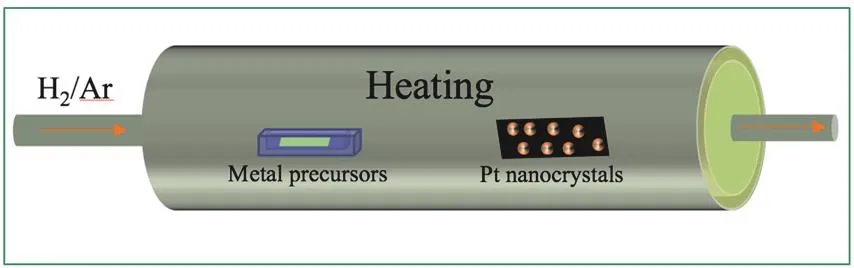
图4 CVD反应示意图Fig. 4 Schematic diagram of CVD.
Komatsu等人63将Pt负载于沸石上(Pt/HZSM-5),随后利用H2将蒸发的乙酰丙酮氯化锗(Ge(acac)2Cl2)沉积到Pt/HZSM-5表面,在550 °C下加热1 h后合成了平均粒径为34 nm的PtGe金属间化合物,而且HZSM-5载体没有发生崩塌。通过表征发现,尽管少量的Ge(acac)2Cl2分子可能分解成了较小尺寸的络合物进入到HZSM-5的孔中,但大部分的Ge(acac)2Cl2分子仍然停留在HZSM-5表面与Pt相互作用,随着温度的升高Ge(acac)2Cl2转化为金属Ge,并扩散进入Pt晶格中逐渐形成金属间化合物相。Saedy等人64使用H2PtCl6为金属前驱体制备出Pt纳米晶,然后使用CVD法,将乙酰丙酮钴(Co(acac)2)作为Co源,将Co沉积在Pt表面,在550 °C下加热6 h后合成了氧化铈纳米棒(NRs)负载的结构稳定有序的Pt3Co纳米晶金属间化合物,通过分子动力学和密度泛函理论(DFT)研究了沉积和合金化的过程,证明了沉积的可行性,发现Co原子优先沉积在Pt表面,而不是氧化铈NRs表面,并通过X射线吸收光谱证明了合金化后Pt和Co的电子结构发生了变化,Co与Pt间通过电子转移形成了Co―Pt键。Co的存在提高了Pt3Co耐CO能力,并使催化剂能够在富氢气氛下优先氧化CO,通过阿伦尼乌斯图证明该方法合成的Pt3Co比浸渍法合成的Pt3Co有更小的CO活化能;高角度环形暗场扫描透射电镜(HAADF-STEM)也表明CVD法是一种极其有效的合成单分散均匀双金属纳米晶的方法。CVD法缩短了退火时间,在过去Pt3Co合金是由PtCo固溶体在500–700 °C退火100 h的苛刻条件下转化而来的64,65。但是相对于直接液相合成法,高温诱导结构转变法来说,目前使用CVD法合成Pt基金属间化合物的难度仍然较大。这可能是因为需要长时间加热才能使第二种金属有效地扩散到Pt晶格中,为了进一步降低CVD法的合成难度,未来可以利用高缺陷密度的Pt纳米晶以促进第二种金属在Pt晶格中扩散。
3 铂基金属间化合物纳米晶的电催化性能
为了降低贵金属的负载并提高催化剂的稳定性,研究者们研究了不同Pt基金属间化合物的电催化性能(表3)。尽管研究者的测试条件不完全一致,但还是可以观察到Pt基金属间化合物的循环稳定性明显优于商业Pt/C催化剂,这归因于Pt基金属间化合物独特的有序性结构。然而,针对电催化活性的比较,争议仍然存在。首先,不同研究者采用的商业Pt/C催化剂型号或生产厂家并不完全相同,性能差异巨大。以ORR为例,Johnson Matthey Co.催化剂的质量活性为0.1 A·mg−1,而HiSPEC 2000催化剂的质量活性却可以达到0.9 A·mg−166,67。根据表3数据,显然并不是所有的Pt基金属间化合物的质量活性都超过了0.9 A·mg−1,因此具体指出商业Pt/C的型号是有意义的。然而,大多数研究者都没有提及所用商业Pt/C催化剂的具体型号或生产厂家。值得注意的是,有的Pt基金属间化合物催化剂的催化性能参数(如质量活性)很低,这是因为研究者并没有将催化剂负载于炭或其他载体材料上,如果加入合适的载体材料,催化剂的性能可能还会有所提高68。此外,部分Pt基金属间化合物中的贵金属含量仍然较高69–71,而贵金属含量低的催化剂性能又不太理想,因此如何进一步降低贵金属的含量的同时,提高催化剂的性能是接下来重要工作。
3.1 电催化还原反应(HER,ORR)
3.1.1 电催化HER
由于能量密度高,燃烧后产物只有水等优势,氢作为一种理想的能源载体近年来受到了广泛关注,电化学析氢反应(HER)被认为是最可行的制氢方法之一。HER反应有两个基元步骤:第一步是一个质子吸附在催化剂表面并得到一个电子(Volmer步骤);第二步可以通过两种不同机理进行,或者两个吸附的氢原子结合(Langmuir机理),或者第一步得到的吸附氢原子与溶液中的游离质子或水分子反应并再得到一个电子(Eley-Rideal机理)。后一种反应路线常常伴随比前一种更高的表观活化能。根据极化曲线计算得到的Tafel系数b可以确定哪种机制占主导地位:当b大约为30 mV·decade−1时为Volmer-Tafel机理(Langmuir机理),而当b约为120 mV·decade−1时为Volmer-Heyrovsky机理(Eley-Rideal机理)72,73。
1949年,Leidheiser等人73发现d金属上发生HER的过电势依赖于催化剂的原子间距:理论计算表明原子间距为2.7 Å (1 Å = 0.1 nm)时可得到最低的过电势。Pt的原子间距为2.774 Å,虽然相比于其他过渡金属元素,显示了最高的电催化HER活性,但是通过加入过渡金属调节Pt的d带中心位置,可以继续优化氢吸附能,从而提高Pt基催化剂HER性能。2015年,Li等人57通过退火处理将MgO包覆的哑铃形fcc-FePt-Fe3O4转化为fct-FePt纳米晶,随后用稀硝酸除去MgO壳。Fe与Pt的d轨道强相互作用提高了fct-FePt的稳定性。对比fcc结构的FePt及商业Pt/C,fct结构FePt展示了更好的酸性(H2SO4) HER稳定性,在−0.3 – 0.9 V电势区间循环10000次后,fct-FePt极化曲线位置基本不变,而商业Pt/C催化剂在10 mA.cm−2的电流密度下电势降低了7 mV。随后将fct-FePt运用到质子交换膜燃料电池中进行测试,其开路电压达到了0.989 V,最大功率密度达到了0.70 W·cm−2,优于商业Pt催化剂的性能(开路电压为0.959 V,最大功率密度为0.63 W·cm−2)。
2018年,Lim等人69使用惰性气体保护的油酸油胺体系,在不同温度下分别制备了Pt3Ge和PtGe2金属间化合物纳米晶。令人惊讶的是,对于酸性HER反应,PtGe2金属间化合物纳米晶几乎无催化活性,而Pt3Ge金属间化合物纳米晶展示了与商业Pt/C相当的活性,在5 cm2的HER活性面积下,Tafel斜率为37 mV·decade−1,证明其发生的是Volmer-Heyrovsky机理。Pt3Ge在酸性HER反应条件下−0.3 V – 0.2 V (vsRHE)的电势区间循环10000次后,极化曲线位置不变,展示了优异的稳定性。2019年,Lim等人68又报道了Pt3Ga金属间化合物纳米晶在酸性HER反应中优异的性能:10 mA.cm−2的电流密度下27 mV的过电势,Tafel斜率为43.3 mV.decade−1(商业Pt黑为47.6 mV.decade−1),10000次循环之后性能保持不变。2019年,Li等人61发现负载于二维过渡金属碳化物Ti3C2Tx上的Pt经550 °C还原生成的Pt/Ti3C2Tx(图5)表现出良好的HER性能:10 mA.cm−2的电流密度下32.7 mV的过电势,Tafel斜率为32.3 mV.decade−1,其质量活性和比活性分别是商业Pt/C的4.4和13倍。此外,PtDy和PtHo金属间化合物70也在碱性HER反应中展示
了比纯Pt更优异的催化活性。
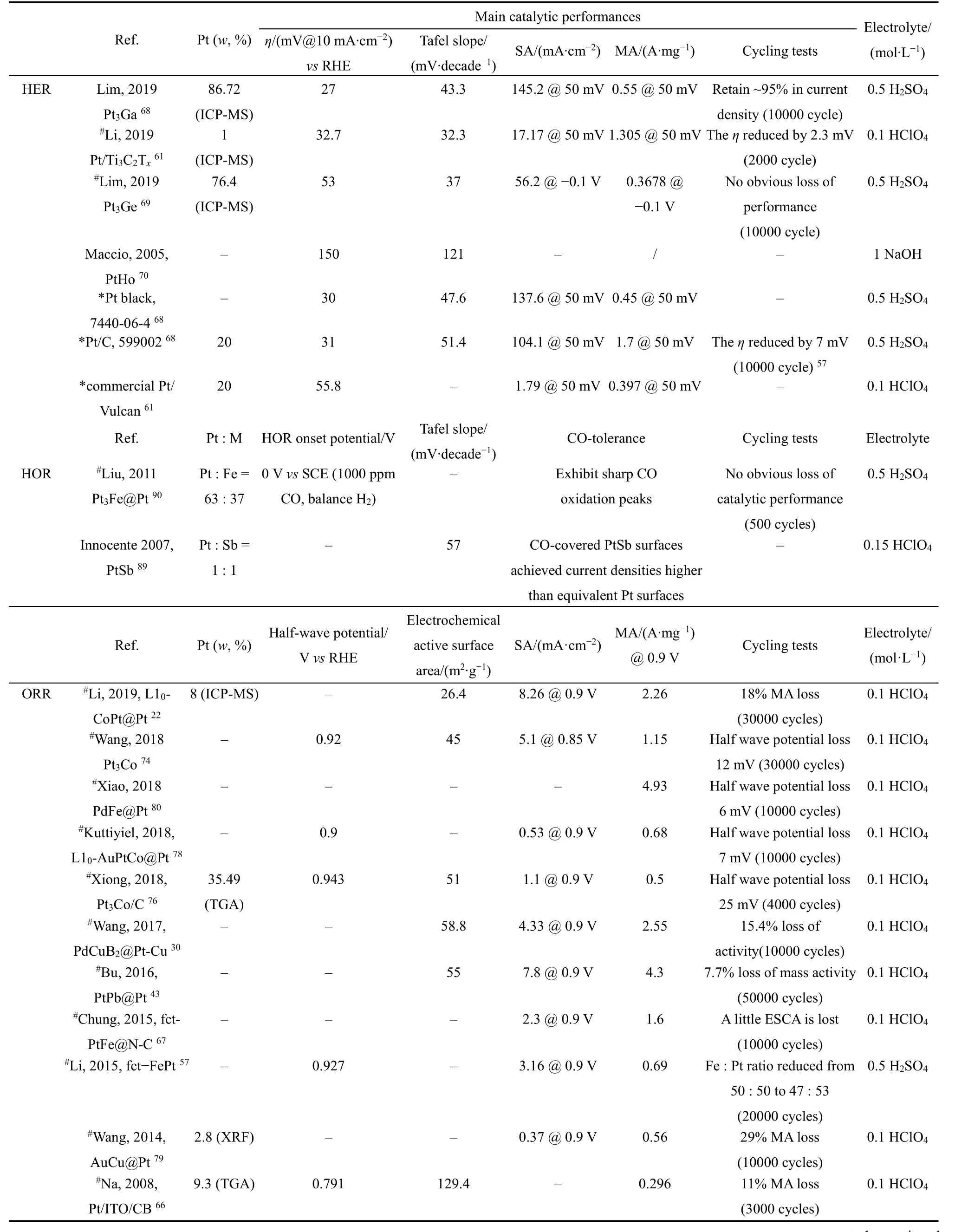
表3 近年在各类电催化反应中性能优异的铂基金属间化合物催化剂的电化学性能Table 3 Electrochemical performance of platinum-based intermetallic catalyst for various electrocatalytic reactions in recent years.
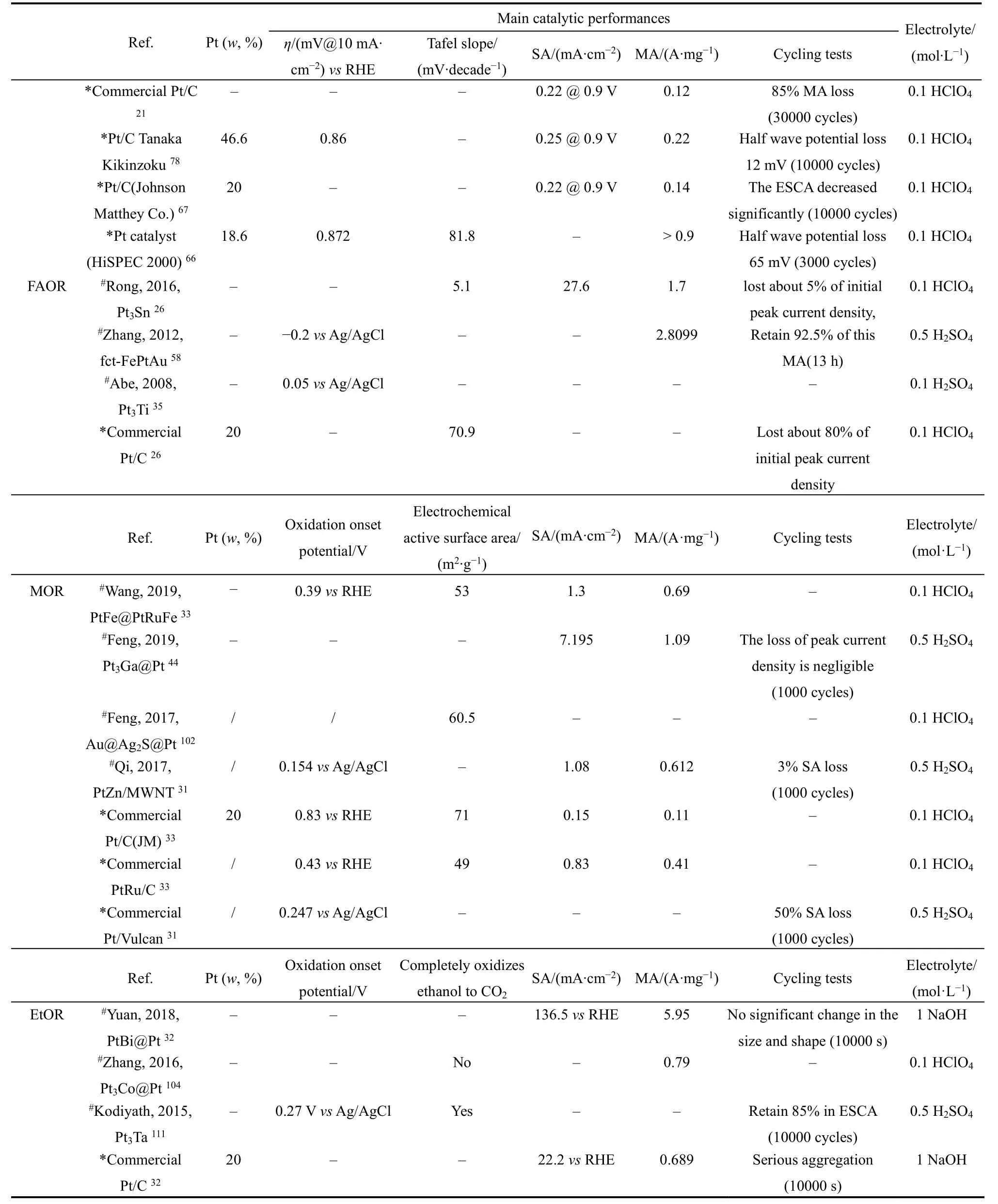
continued Table 3
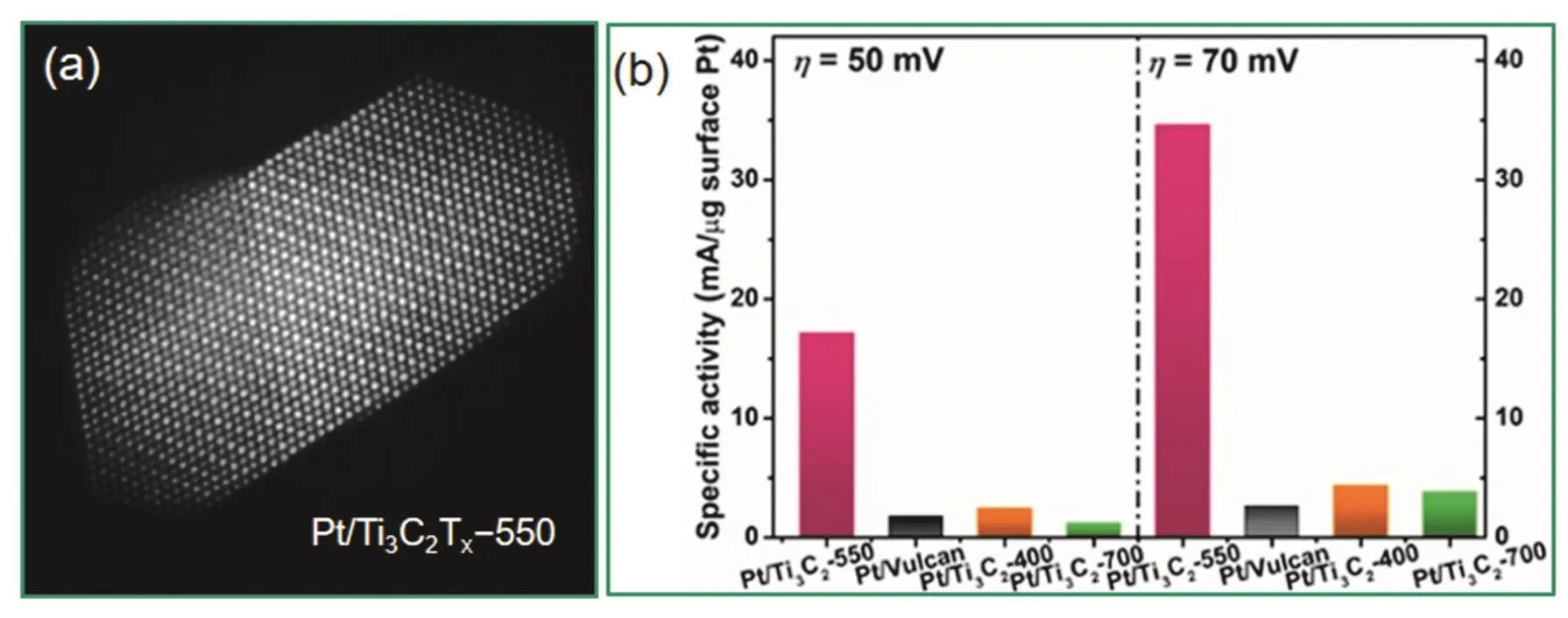
图5 (a) Pt/Ti3C2Tx-550的AC-HAADF-STEM图(b)不同还原温度下的Pt/Ti3C2Tx及商业Pt/C的HER比活性61Fig. 5 (a) AC-HAADF-STEM image of Pt/Ti3C2Tx-550, (b) specific activity of the Pt/Vulcan and Pt/Ti3C2Tx catalysts for HER 61.
3.1.2 电催化ORR
低成本、高活性和催化性能稳定的催化剂是燃料电池大规模应用的关键。燃料电池中,通常阴极反应速率小于阳极反应速率;由阴阳两极反应速率不等造成的活化极化使燃料电池的电动势低于可逆电动势,并使电池功率密度下降。为了减小活化极化的影响,提高电池功率密度,一个重要策略就是提高阴极ORR的反应速率(提高电流密度)。因此,ORR是目前电化学反应中研究最多的。由于ORR的发生需要很高的电势(1.23 VvsSHE),在如此高的电势下,很多Pt基合金催化剂的稳定性很难维持。美国能源部2020年质子交换膜燃料电池(PEMFC)中的技术目标(表1):质量活性达到0.44 mA.cm−2且在900 mV下经过30000次循环后,初始质量活性的下降比例要小于40%9。目前,科学家们开发出了多种策略提高Pt基催化剂的性能。
2018年,Wang等人74利用Pt和Co两种元素,通过在金属−有机骨架(MOF)衍生的碳上对铂纳米晶进行简单地热处理,使原子分散的Co位点嵌入到MOF衍生的碳中,随后扩散到Pt纳米晶中,形成有序的Pt3Co金属间化合物结构。此种Pt3Co纳米晶有着优异的ORR活性和稳定性,在0.6–1.0 V之间的30000次电势循环后半波电势仅损失12 mV。一方面,Co和Pt之间的高度有序性和强烈的d轨道相互作用不仅稳定了Pt,也抑制了Co在酸性溶液中的溶解。另一方面有机骨架可能改变了Pt3Co的电子结构,加强了载体与Pt3Co的作用,有效地抑制了纳米晶的聚集,从而提高了催化剂的稳定性。

图6 氧吸附能的DFT计算43Fig. 6 DFT calculations of oxygen adsorption energyy 43.
利用金属间化合物超高的结构稳定性,设计其为催化剂内核,金属Pt或非金属为壳层材料,由于核壳结构之间的晶格失配导致的表面应变(压缩应变及拉伸应变)是提高ORR性能的重要策略之一2,75,77。2015年,Chung等人67对聚多巴胺包覆的fcc结构PtFe纳米晶进行退火处理,获得了含氮炭壳包覆的fct结构PtFe金属间化合物纳米晶。即使在燃料电池苛刻的反应条件下,这种原位形成的炭壳仍然能有效的抑制纳米晶的聚集和溶解,使得该催化剂拥有出色的ORR稳定性,在持续100 h的膜电极组件中测试,最大功率密度仅损失3.4%。2016年,Bu等人43报道的PtPb@Pt核壳纳米片以金属间化合物PtPb为核,纯Pt为壳,展示了显著的双轴应变:在纳米片结构的上界面,Pt[011]方向11%的压缩应变,在Pt[100]方向7.5%的拉伸应变;在纳米片结构的侧界面,Pt[001]方向7.5%的拉伸应变,在Pt[110]方向1.0%的压缩应变。双轴应变导致PtPb@Pt纳米片优异的ORR性能:0.9 V (vsRHE)比活性和质量活性分别为7.8 mA·cm−2和4.3 A·mg−1,50000次循环后活性无明显衰减。在电化学测试下,精心设计的铂壳可以通过位置交换机制阻止内部过渡金属的损失,从而提高其ORR稳定性。研究者对PtPb六方纳米片上的氧吸附能(EO)进行了DFT研究(图6)。已知ORR反应在EO的最优值处达到最大活性,为了研究方便,研究者将最优EO值设为0 eV,并使用ΔEO表示给定EO值相对于最优值的差值。在Pt(110)晶面上的ΔEO是在Pt[110]和Pt[001]方向上应变的函数,图6b,d中横纵坐标中的负和正的百分数分别代表压缩和拉伸应变,阴影部分代表双轴应变位于这些位置的Pt(110)会显示比平的Pt(111)更高的活性。b,c,d图分别代表Pt(110)晶面上三个最稳定的氧气吸附位点上的双轴应变与ΔEO的关系:凹位点(图6a中h位置)、Pt[001]方向桥位点(图6a中b1位置)和Pt[110]方向的桥位点(图6a中b2位置)。由图可知,在纳米片结构的侧界面Pt[001]方向7.5%的拉伸应变,在Pt[110]方向1.0%的压缩应变的情况下,h位点稳定且ORR活性高,而b1位点活性低;Pt[001]方向上7.5%的拉伸应变可以削弱b2位点上原本太强的Pt―O键,使得PtPb六方纳米片的上下面及侧面的b2位点上氧吸附能得到优化,从而提高其ORR活性。2019年,Li等人22合成出了一种强铁磁性带有2–3 Pt原子壳层和金属间化合物内核的L10-CoPt/Pt 纳米晶,这种材料在80 °C膜电极反应条件下质量活性初始为0.56 A.mg−1,30000次循环后为0.45 A.mg−1(下降了19.6%),远远高于美国能源部2020年的技术目标。薄Pt壳层增加了ORR反应的活性,Co原子的配体效应和Pt壳层的双轴应变削弱了中间产物与表面Pt原子的结合,降低了反应过电势;内核L10结构可保护Co原子尽量少地被腐蚀以保持催化剂的活性,从而使L10-CoPt/Pt纳米晶的ORR性能得到大幅度提高。多次催化循环反应后表征发现,金属间化合物L10-CoPt的核结构能够被很好的保留,使得催化剂粒子仍能保持均匀的尺寸和较好的分散性,这是该催化剂具有超高稳定性的原因。依据表面原子排列对ORR动力学的重要作用,Kuttiyiel等人78设计并合成了L10-AuPtCo金属间化合物和被薄层Pt壳包覆的有序L10-AuPtCo金属间化合物(L10-AuPtCo@Pt),二者催化ORR的质量活性分别达到0.49和0.68 A·mg−1;通过DFT计算,发现L10-AuPtCo@Pt结构中Au趋向于迁移到L10-AuPtCo@Pt的表面,并优先填充纳米晶的角点和棱边,从而抑制了Pt的氧化和Co的溶解,大大提高了纳米晶的稳定性。

图7 (a) PdCu B2@Pt-Cu和Pt-Cu两种纳米颗粒ORR比活性和质量活性对比及(b) PdCu B2@Pt-Cu的AC-HAADF-STEM表征20Fig. 7 (a) Specific activity and mass activity of PdCu B2@Pt-Cu and Pt-Cu (b) AC-HAADF-STEM images of PdCu B2@Pt-Cu 20.
2017年,Wang等人20在金属间化合物PdCu B2(CsCl结构,图7)纳米晶核上沉积fcc结构Pt-Cu合金壳层,得到具有高表面应变的PdCu B2@Pt-Cu纳米晶。由于核和壳层之间存在较大结构和晶格上的失配,Pt-Cu覆盖层表现出与方向有关的表面应变,具体表现为施加在Pt-Cu[111]上的压缩应变比Pt-Cu [200]上的压缩应变小。这种具有高度表面应变的PdCu B2@Pt-Cu纳米晶的ORR比活性(4.33 mA·cm−2)和质量活性(2.55 A·mg−1)分别是Pt-Cu纳米晶的4.6和8.7倍,PdCu B2核中有序的金属间化合物结构和强键合作用使其在酸性环境中具有良好的抗溶性,催化剂具有优异的稳定性。Wang等人79通过DFT计算发现1.5个单层Pt包覆的AuCu(111)表面氧吸附能比Pt(111)低0.2 eV,这是理论上催化ORR的最佳表面氧吸附能值。随后,研究者用Pt将AuCu金属间化合物纳米晶表面的Cu置换,成功制备了壳层厚度小于2个原子层的Pt包覆AuCu催化剂,其对ORR的表面催化活性(0.37 mA·cm−2)是商用Pt/C催化剂的两倍以上,对ORR的质量活性达到了0.56 A·mg−1@ 0.9 V。与之类似,2018年,Xiao等人80利用浸渍还原法制备出了PdFe/C,进一步采用自发置换的方法获得了fct结构的PdFe@Pt/C,其质量活性为4.93 A·mg−1DFT计算表明PdFe@Pt(111)表面氧吸附能比Pt(111)低0.16 eV,接近理想的0.2 eV。鉴于其优异的ORR能力,研究者将其作为阴极,锌箔作为阳极制备了锌空气电池,并获得了优异的性能,峰值功率密度达到了293 mW·cm−2,峰值电流密度达到了342 mA·cm−2。
综上所述,调节表面应变以减小氧吸附物种与Pt的成键强度是提升ORR性能的关键因素之一。
不仅应变工程,电子转移和几何效应也使部分铂基复合催化剂,如铂基氧化物催化剂,在ORR反应中体现出良好的性能。Na等人66提出了一种炭黑负载的Pt/铟锡氧化物(ITO)三维结构,其Pt含量为9.3% (w),较小尺寸的Pt (1.9 nm)和ITO (5.6 nm)是ORR质量活性(191 mA.mg−1)显著优于商业催化剂(HiSPEC 2000,其Pt含量为18.6% (w),质量活性为116 mA·mg−1)的原因。在经过3000次循环加速耐久性测试(ADT)后,催化剂的电化学表面积保留率高达74.9%。此外,在电化学反应中,ITO的存在可能会去除吸附的CO物种,因此在3000次ADT后,催化剂的CO氧化峰仅位移0.05 V (商业催化剂的CO氧化峰位移0.14 V),表现出了较强的CO耐受性。此外,通过减小ORR的过电势从而提高燃料电池的开路电压,也是提高燃料电池功率密度的重要方法。Masuda等人81使用沉淀法和共浸渍法合成Pt-CeOx/C氧化物复合催化剂,该催化剂的ORR起始电势比商业铂炭(Johnson-Matthey)高50–80 mV,即该催化剂有更小的过电势。对催化剂的PtL3和CeL3边及商业铂炭的PtL3边进行X射线吸收精细结构谱测试,其结果表明ORR活性的增强是由于CeOx层抑制了氧化铂的形成,Ce3+向Pt转移了部分电子,抑制了Pt的氧化。Sasaki等人82在碳负载的氧化铌纳米晶(NbO2或Nb2O5)上生长少量单层铂,氧化铌纳米晶不仅发挥载体的作用防止铂团聚,其表面大量存在的OH或O的排斥作用使得Pt表面吸附的OH物种减少,氧空位的存在还使得O―O键更容易断裂,因此Pt/NbO2/C催化剂在ORR反应中表现出良好的性能,其比活性为4.9 mA·cm−2,质量活性为0.21 A·mg−1,并且在30000次循环后仍表现出较好的抗铂溶解稳定性,其半波电势仅下降23 mV;相比之下,商业Pt/C的半波电势下降了40 mV。是否所有氧化物与Pt都可发挥类似协同作用,作用强弱的比较是铂基氧化物复合催化剂值得被深入研究和探讨的重要课题。
由于ORR反应的重要性,已经有大量系统而深入的综述发表。2015年,Wang等人83综述了碳支持的Pt基合金材料的颗粒尺寸、形状和组成对其在PEMFC中ORR性能的影响。2016年,Strasser等人71综述了通过去合金法得到的Pt基核壳结构催化剂的ORR性能。2017年,Luo等人84综述了多金属金属间化合物纳米晶在能量转化相关的电催化反应中的应用;同年,Yan等人36综述了金属间化合物的合成与催化应用;Antolini85对比了Pt基和Pd基合金与金属间化合物在ORR及低温燃料电池中的性能。2018年,Xiao等人15综述了用于电催化的金属间化合物纳米晶的最新进展;同年,Liang等人86综述了通过调节晶体结构提升Pt基金属间化合物催化性能的工作;Gamler等人47也对比了合金与金属间化合物纳米晶在电催化反应中的性能。2019年,Li等人16综述了金属间化合物的可控合成与增强的电催化性能;Rößner等人72全面地综述了金属间化合物催化的电化学能量转化过程。这些综述从不同的角度对ORR工作进行了总结与评述,为此领域的科研工作者提供了指导。
3.2 电催化氧化反应(HOR,FAOR,MOR,EtOR)
3.2.1 电催化HOR
氢气氧化反应(HOR)与HER类似,反应过程需两步,或者通过Volmer-Tafel机理(需要两个相邻活性位点)或者通过Volmer-Heyrovsky机理(仅需一个吸附位点)。氢吸附能是影响此反应活性的关键因素,可通过电子效应调节催化剂的d带中心从而影响氢吸附能72。如前所述,与阴极ORR相比,酸性条件下的HOR反应动力学比传质快很多,很少量的Pt负载量(0.05 mg·cm−2)就可满足商业应用需要。然而碱性HOR活性却比酸性环境的低两个数量级,原因可能是其他阳离子的竞争吸附87。同时,设计HOR催化剂的同时需要考虑CO的毒化,这是由于电解液中CO浓度达到10 ppm (1 ppm = 1 ×10−6,体积分数)时即可毒化Pt阳极88。有些比Pt便宜的金属可以在更低的电势吸附OH物种,降低CO的氧化电势,让Pt与这样的金属形成合金或金属间化合物是抑制Pt失活的有效策略。利用铂基金属间化合物的配体效应(Pt与邻近原子之间电荷转移)和几何效应(应变表层),可有效增加Pt位点上OH的吸附强度。
Innocente等人89研究表明金属间化合物PtSb和PtSn (均为NiAs型晶体结构)在高氯酸溶液中对CO分子的亲和力弱于纯Pt,因此表现出更好的酸性HOR性能。Santos等人25通过对铂锡体系中不同金属间化合物的模拟计算和实验研究发现,不同铂锡比例的金属间化合物对HOR的催化活性有显著不同:Pt3Sn (Cu3Au型晶体结构)和PtSn(NiAs型晶体结构)在催化HOR时展示了与纯Pt相当的活性,而PtSn2(CaF2型晶体结构)无催化活性。氢的电催化作用取决于Pt的d带和氢的1s轨道耦合,利用迭代方法求解密度泛函理论的Kohn-Sham方程,计算表明,以上三种金属间化合物的电子性质差别较小,Pt的d带都位于费米能级以下,这说明电子由Sn流向了Pt,并使Pt的d带全部被填满。随后研究者通过理论计算得到了Tafel反应的活化势垒,计算结果表明活化势垒在0.3–0.5 eV时对反应是有利的,但是PtSn2表面的活化势垒过高(0.65 eV),导致PtSn2催化剂无HOR催化活性。Liu等人90研究了Pt3Fe合金、金属间化合物纳米晶以及金属间化合物为核的Pt3Fe@Pt颗粒的HOR活性及CO耐受性。利用旋转圆盘电极测量H2+ CO (1000 ppm)混合物的电氧化极化曲线,发现HOR活性的大小为:Pt3Fe合金(−0.2 VvsSCE (饱和甘汞电极)) > Pt3Fe@Pt (0 VvsSCE) > 纯Pt > Pt3Fe金属间化合物。随后测定了催化剂在纯CO环境下的循环伏安曲线,发现Pt3Fe金属基化合物几乎显示不出CO氧化峰。也就是说,Pt3Fe金属间化合物纳米晶的HOR活性和耐CO中毒能力都比纯Pt差,研究者认为这可能是由于Pt3Fe金属间化合物纳米晶表层富Fe而暴露的Pt位点过少引起的。
3.2.2 电催化FAOR
甲酸氧化反应(FAOR)是一种双电子转移反应,可以通过三种不同的反应路径进行27,91。其中,直接路线(脱氢反应)是最有利的路线,因为只包含一个基元反应,优化此类催化剂的方法就是抑制甲酸和CO分子在催化剂表面的吸附。两种间接路线是通过OH*氧化CO的方式,在HOR部分已经着重讨论,Pt基催化剂可以利用其他金属对OH*的吸附,在提高反应速率的同时可以减弱Pt的中毒。
Abe等人35比较了Pt3Ti合金、金属间化合物纳米晶,商业纯Pt/C和Pt-Ru/C纳米晶的FAOR和MOR性能。与Pt-Ru/C (0.14 VvsAg/AgCl)和Pt/C(0.40 VvsAg/AgCl)相比,Pt3Ti合金(0.04 VvsAg/AgCl)和金属间化合物(Cu3Au型晶体结构,0.05 VvsAg/AgCl)纳米晶均具有较低的氧化起始电势,并且金属间化合物纳米晶表现了0.34 mA.cm−2@ 0.6 V电流密度。研究结果表明,Pt3Ti金属间化合物催化FAOR反应时直接路线占优势,CO吸附强度降低,这是催化剂活性增加的原因。
具有高比表面积、良好导电性的催化剂载体可以改善催化剂的化学环境提升催化剂的稳定性,是提升催化剂性能的有效策略。Xu等人92合成了氮掺杂石墨烯(NG)支撑的PtAu金属间化合物核/Pt枝晶壳纳米晶(PtAu/Pt)催化剂。一方面,作为优秀的载体,NG的引入促进了载体内的电子传输,增强了纳米晶与载体之间的作用,提高了催化剂的稳定性;另一方面,树枝状的Pt壳可以暴露出更多甲酸分子可用的表面活性区域。因此该催化剂表现了出色的质量活性(1847.1 mA·mg−1),比活性(14.95 mA·cm−2)和稳定性,在循环500次的情况下几乎没有活性衰减。利用惰性金属(如Au)对金属间化合物保护是另一重要策略。Zhang等人58发现fct-FePtAu在FAOR中的质量活性高达2809.9 mA·mg−1,在13 h稳定性实验测试后仍能保持92.5%的活性。Au的引入和偏析促进了其中有序fct结构的形成,而不存在Au或fct结构的Pt,FePt和fcc-FePtAu纳米晶等催化剂均易产生CO中毒和酸溶现象,因此,Au在纳米晶表面的富集是导致FAOR超高活性和稳定性的可能原因。此外,在FAOR中表现出良好活性的金属间化合物还有PtZn,Pt3Zn93,Pt3Zr94,PtCd95,PtBi96,97,PtPb98和Pt47Ag5399;值得注意的是,Pt3Mn(Cu3Au型晶体结构)纳米立方体表现了比商业Pt/C差的FAOR性能100,这可能是由于退火过程中,纳米材料表面吸附了少量分解的含碳物质101,从而减少了催化剂暴露的活性位点数量。
3.2.3 电催化MOR
甲醇氧化反应(MOR)是直接甲醇燃料电池(DMFC)对应的阳极反应,如上文所述,目前基础实验室研究中一些对于ORR反应(DMFC的阴极反应)的研究成果基本达到了美国能源部的目标,所以MOR的活性和稳定性的提升更迫在眉睫。与前面提到的其他氧化反应相比,MOR是6个电子转移的过程,必须经历多步反应路径,反应中间体也比较多样。上面提到的甲酸也是可能的中间体,因此Casado-Rivera等人98曾总结对MOR表现出活性的金属间化合物也必然能催化FAOR。然而,在MOR反应中,由于中间体更多样,对FAOR有优异催化性能的催化剂并不一定会有优异的MOR性能。此外,MOR反应中CO的吸附更为普遍,因此相比于FAOR,通过双功能或溢流机理为CO氧化提供OH*在MOR中显得更为重要72。
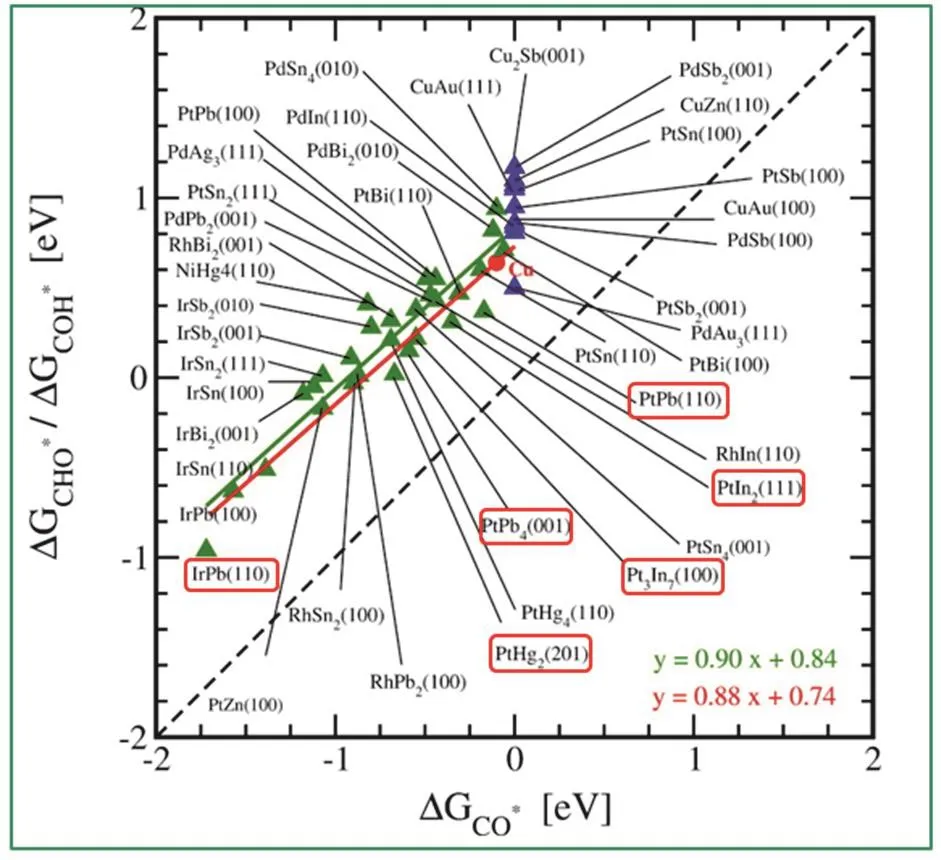
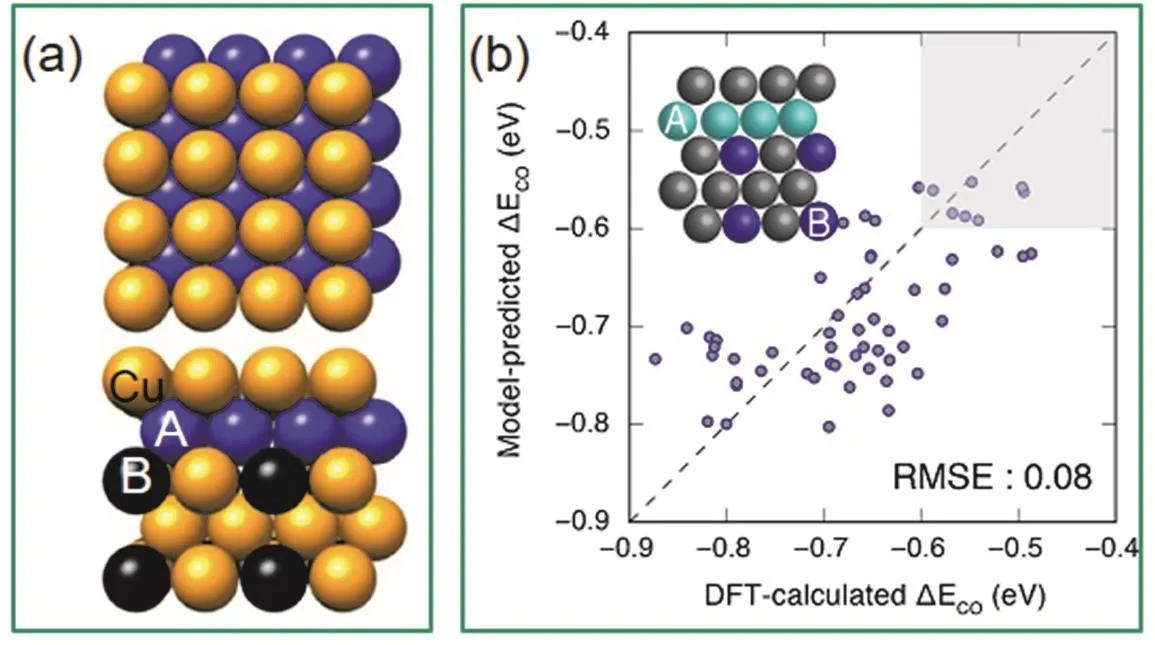
Wang等人33结合空间限域效应和表面工程策略,制备了核-壳结构PtFe@PtRuFe纳米催化剂,其具有稳定的金属间化合物PtFe核及3–5原子层厚的PtRuFe壳结构。由于电子由Fe/Ru向Pt的转移,减弱了Pt-COads吸附能,从而降低了CO的中毒现象,提高了催化剂的稳定性。Feng等人102使用晶种诱导生长法合成了核-壳-壳Au@Ag2S@Pt纳米复合材料,这种结构的材料在阳极MOR中有着优异的性能,将其组装为DMFC,在甲醇浓度为10 mol·L−1时获得了89.7 mW·cm−2的功率密度,而商业DFMC仅为40.5 mW·cm−2。2017年,Qi等人31报道了多壁碳纳米管(MWNT)上PtZn纳米晶(3.2 ± 0.4 nm)使MOR体系经历“非CO”反应路径表现出优异的活性及抗中毒性;其原因为OH*与Zn原子的结合强度高于Pt原子,使得PtZn表面的(CH2O* →CH2O* + OH* → H2COOH*)路径取代了纯Pt表面的(CH2O* → CHO* → CO*)路径,后一路径常会导致Pt催化剂失活(图8)。DFT计算探究了在PtZn和Pt材料上MOR反应机理以及粒径大小和金属组成对催化过程的影响,小的纳米晶通常比大的纳米晶具有更高的棱边和顶角比例。在这项工作的理论计算过程中使用PtZn(111)、PtZn(211)和Pt24Zn24簇分别代表台阶、棱边和顶角,而计算结果MOR的表观活化势垒由小到大顺序为Pt24Zn24 图8 PtZn金属间化合物纳米颗粒的(a) HAADF-STEM图像(标尺:1 nm)和(b)不同晶面MOR过程的反应机制31Fig. 8 (a) HAADF-STEM images of PtZn intermetallic NPs, scale bar, 1 nm, and (b) calculated reaction mechanisms of MOR process on different crystal plane 31. 3.2.4 电催化EtOR 乙醇具有较高的能量密度,EtOR常有两个可能的路径:完全氧化时有12个电子转移(CH3CH2OH + 3H2O = 2CO2+ 12H++ 12e−),过程中要断裂C―C键,产物为二氧化碳;不完全氧化时,C―C键不断裂,产物为乙酸(CH3CH2OH + H2O =CH3COOH + 4H++ 4e−)或 乙 醛 (CH3CH2OH =CH3CHO + 2H++ 2e−)104,不完全氧化时,转移电子数低于12个电子,能量无法得到完全利用。因此,提升催化剂在EtOR反应中断裂C―C键的性能是提高该反应效率的关键思路之一。与MOR,FAOR催化剂设计思路一致,为了使Pt催化剂上的EtOR顺利进行,需要引入一种更亲氧的元素形成铂基金属间化合物,通过双功能机制促进C―C键断裂,亲氧元素提供高活性的*OH用于生成C―O键105。 Casado-Rivera等人98比较了一系列Pt基金属间化合物的FAOR,MOR和EtOR催化性能。PtBi2,Pt2Sn3和Pt3Sn无EtOR活性,有趣的是,在MOR中无活性的PtBi和PtMn却可以催化EtOR。除了PtBi,PtPb和PtIn的EtOR峰电流密度都高于纯Pt。然而,所研究的四种Pt基金属间化合物都无法断裂C―C键,将乙醇完全氧化为CO2,而是停在了乙酸这一步;将底物换为乙酸的电催化实验也证明了这四种Pt基金属间化合物对乙酸氧化没有催化活性。Herranz等人106利用原位红外反射吸收光谱(IRRAS)和微分电化学质谱方法研究了Pt3Sn和铂黑的EtOR性能,结果表明Sn的加入促进了COad在较低电势(200 mV)处的氧化。然而,Pt3Sn比铂黑更难使C―C键断裂生成CO2,Pt3Sn的催化产物主要为乙醛和乙酸。Kwak等人107也证明了Pt3Sn有比Pt/C更高的EtOR活性,然而他们没有分析反应产物。Ramesh等人108发现Nb的加入也可以促进Pt的EtOR和FAOR性能。Sun等人109发现将C+溅射在PtPb纳米片表面造成的缺陷改善了催化剂对多种电催化反应(MOR, EtOR和ORR)活性。此外,核壳结构及应变工程策略也在EtOR中得到了广泛应用。Gunji等人110发现Pt3Pb@PtPb有比纯Pt3Pb,纯PtPb,商业Pt-Ru和Pd/C更高的碱性MOR及EtOR活性。Zhang等人104发现Pt3Co@Pt (0.79 A·mg−1)在EtOR反应中的质量活性是商业Pt/C (0.32 A.mg−1)的2.5倍,通过原位红外光谱测试(FT-IRS)分析发现,Pt3Co@Pt催化产物中乙酸的含量要高于CO2,即乙醇的部分氧化过程比C―C键断裂更易发生;他们通过理论计算研究了Pt壳层结构的阶梯状Pt3Co(211)晶面上的EtOR电氧化反应机理和路径。计算结果表明,在此阶梯状晶面上CH3CHOH*通过α-C―H键断裂最终形成乙酸的路径需克服的最高反应势垒为0.16 eV,CH3CHOH*的O―H键断裂形成CH3CO*为决速步;而通过CH3CHOH*的β-C―H键断裂生成二氧化碳的路径需克服的最高反应势垒为0.72 eV,CH2CHOH*,的C―H键断裂生成CH2COH*为决速步。相比之下,显然Pt3Co@Pt在EtOR反应中发生第一种反应路径的占比更大。Yuan等32研究者们采用了X射线光电子能谱和X射线吸收近边结构测试,证实了在PtBi@Pt纳米片中,电子会从铋转移到铂上,形成富电子铂。Pt位点周围电子密度的增加会填充部分Pt 5d能带;当d带中心向下移动时,会促使表面含氧物质如吸附的中间体COad减少,从而使吸附乙醇分子的活性位点增多,EtOR催化活性增强。同时表面Pt薄层与内部结构的晶格失配和收缩而表现出的应变效应,调整了催化剂的电子和几何结构,该催化剂体现出了良好的碱性EtOR催化性能,通过TEM对循环后的几种催化剂进行观察发现,Pt/C已经发生了明显的团聚,而金属间化合物结构使得PtBi@Pt催化剂仍以良好的尺寸和分散状态存在。目前,由于C―C键极性较低、断裂难度高,可以完全将乙醇氧化为CO2的Pt基金属间化合物还比较少,这是因为,乙醇氧化的中间产物CO会严重地毒化催化剂。Kodiyath等人111将Pt粉和钽(Ta)粉混合在1000 °C真空条件下退火72 h制备了TaPt3金属间化合物。IRRAS分析发现TaPt3可以高效地氧化乙醇为CO2,将其组装为直接甲醇燃料电池进行测试,在1.6 mA·cm−2的电流密度下获得了0.215 mW的功率密度。Ta元素属于前d区金属相比后d区金属来说,更具电正性和亲氧性,更有利于*OH的形成,因此促进了乙醇的完全电氧化。 由于催化剂中Pt和Pd等贵金属普遍储量少、价格高,近几年含有少量贵金属Pt的催化剂成为降低燃料电池成本的突破口,研究者们找寻合适的非贵金属元素和Pt/非贵金属比例以合成含少量Pt的金属间化合物作为高效催化剂,过渡金属碳化物因其催化性能与铂族金属相似,且具有较高的CO耐受性而备受关注。Nia等人112合成了碳化钨负载铂(3% (w))金(3% (w))锡(10% (w))催化剂(PtAuSn/W2C),这种低贵金属负载量催化剂在70 °C下催化的直接乙醇燃料电池EtOR反应具有超低 的乙醇氧化起始电压和超高的CO2转化效率。研究采用了电化学质谱(EC-MS)和FT-IRS等原位技术分析反应过程和机理,0.65 V (vsRHE)下在线定量EC-MS检测到的CO2转换效率为6.5%,FTIRS结果表明在电势为0.05 V (vsRHE)时C―C键断裂,EtOR性能在高达70 °C下性能仍然有大幅度提升的原因可能是,W2C载体向所有金属粒子的电荷转移,提高了金属粒子的电子密度,从而提高了催化剂的耐CO中毒能力。通过精确的调整合成参数,引入非金属元素也是合成低Pt催化剂的重用手段。Xue等人113将H2PtCl6·6H2O,SnCl2·2H2O溶液与碳黑(Vulcan XC-72)搅拌混合,加入次磷酸钠(NaH2PO2·H2O)和柠檬酸钠(Na3C6H5O7·H2O)试剂后,加入氢氧化钠溶液调节pH,然后在100 °C下反应一段时间后,获得了粒径为2 nm的Pt3Sn1P2/C催化剂。与用乙二醇还原法和硼氢化还原法制备的两种PtSn/C催化剂相比,该催化剂具有更高的EtOR活性,用于直接乙醇燃料电池(70 °C)时获得了0.797 V的开路电压和61 mW·cm−2的功率密度。通过XRD和TEM表征证明磷(P)的引入有效抑制了PtSn的聚集,并显著减小了PtSn的粒径,从而提高了催化剂性能。 传统催化剂的优化设计主要是依赖于经验,而量子化学和计算机的快速发展,从理论上为催化剂的优化设计提供了支持114–117。 金属间化合物的有序结构对催化剂的选择性会产生重要影响。针对电催化CO和CO2还原过程中,高附加值产物的选择性差的问题,Karamad等人17模拟了金属间化合物的表面,它们由对HER活性差的元素包围的对CO还原活性高的单原子中心组成。可以还原CO的过渡金属元素包括Ru,Co,Rh,Ir,Ni,Pd,Pt和Cu,对HER催化性能不好的金属元素包括Ag,Au,Cd,Zn,Hg,In,Sn,Pb,Sb和Bi。利用DFT的方法,计算得出34种稳定的二元金属间化合物表面,通过调控活性中心的尺寸和组成它们的元素可以优化其电子和几何性质。由于在已有的所有过渡金属表面,CO质子化为*COH或*CHO是决速步,因此他们假设这一个步骤为CO还原的决速步骤。他们将*CHO或*COH的自由能与相应的*CO自由能绘制出来(图9),于是图上每个点代表的催化剂的过电势就可以用该点到对角线的距离表示。结果表明有6个金属间化合物面具有对CO还原的选择性,其中PtIn2(111)和PtHg2(201)甚至表现出比已有最好的CO还原催化剂Cu更低的还原过电势。 图9 将*CHO或*COH的自由结合能与*CO的相应能作图17Fig. 9 Free binding energies of *CHO or *COH are plotted against the corresponding energies for *CO 17. 图10 (a) Cu3B-A@CuML结构,黄色:Cu原子,黑色:原子B,蓝色:原子A。(b)基于机器学习模型计算的Cu3X-Ni@Cu和Cu3X-Rh@Cu合金(X:过渡金属)的CO吸附能比较119Fig. 10 (a) Structures of {100}-terminated Cu3B-A@CuM alloy models used in this study. (b) the parity plot shows a comparison of the CO adsorption energies on the Cu3X-Ni@CuML and the Cu3X-Rh@CuML alloys (X: transition metal atoms) calculated using the machine-learning model with the geometry-based primary features and the self-consistent DFT 119. 伴随近年来机器学习的快速发展,研究者采用针对由不同元素排列组合形成的金属间化合物的全自动筛选方法,使用机器学习和组合优化来指导DFT计算过程,进而预测电催化剂的性能,筛选出性能优异的催化剂,提高实验尝试的成功率118。2016年,Li119等人开发了用于快速筛选过渡金属催化剂的机器学习(ML)模型,将易于获取的表面金属原子的特征—例如吸附位点的局部电负性和有效配位数—数值化呈现。这些特征取决于吸附位点的周围环境,以及活性金属原子的固有性质(如电负性和离子势)。他们以*CO吸附能为重要指标,利用优化后的模型筛选用于电催化CO2还原的暴露(100)晶面的多金属Cu基合金催化剂,结果表明,一些Cu3B-A@CuML合金,包括Cu3YNi@CuML,Cu3Sc-Ni@CuML,Cu3Ti-Rh@CuML,Cu3V-Rh@CuML和Cu3Mo-Rh@CuML具有理想的CO吸附能(图10)。2017年,该课题组又基于量子化学模拟和动力学分析提出了一个系统的机器学习机制用于快速筛选双金属催化剂用于MOR120,重点探讨了甲醇氧化的间接机制,建立了一个催化剂数据库用于优化模型的结构和权重参数,该数据库包含DFT计算得出的在暴露晶面的模型合金表面上*CO和*OH的吸附能以及活性位点的指纹特征。使用已有的约1000个理想合金表面数据集训练的模型可以采集吸附物与金属之间复杂而非线性的相互作用,发现除了Pt/Ru合金外,Pt/Fe、Pt/Co、Pt/Ni可以有效降低MOR反应的过电势,是潜在的MOR催化剂,为更进一步的高级量子计算或实验测试缩小了范围。需要指出的是,机器学习并不是为了取代DFT计算,而是为了基于DFT计算快速筛选催化剂115。Tran等人121针对CO2还原反应和HER,对由31种元素(Ga,Ag,Zn,Sn,Al,Au,Cu,N,V,Ge,Pd,Pt,Mo,Rh,W,Ru,Ni,Re,Os,Ir,Ti,Si,Fe,Pb,As,H,Mn,Sb,Co,In,Cr)组成的1499种金属间化合物晶体催化剂的1684908个吸附位点进行了筛选。研究者建立了一个可以自动生成并存储DFT数据的机制,并将该机制与计算和管理软件以及机器学习模型结合,从而实现了自动化、系统化的DFT计算。为了将吸附位点数值化呈现,研究者考虑了与吸附物(CO或H)配位元素的指纹特征,包括元素的原子序数,元素的Pauling电负性,与吸附物配位元素的原子数量以及吸附物与纯元素之间的吸附能。该模型能够对众多的吸附位点进行快速的分析,并寻找到了54种针对CO2还原反应的催化剂和102种针对HER的催化剂。但是,该模型并没有考虑结构弛豫的影响,因此会产生一定的误差115,目前还处于探索阶段。 Pt基金属间化合物纳米晶作为一类高效稳定的电催化剂,由于其独特的几何效应和电子效应,受到了越来越多的关注。本文评述了近些年来铂基金属间化合物纳米晶的研究进展,重点探讨了三种可控合成方法(直接液相合成法、高温诱导结构转变和化学气相沉积法),以及其在电化学能量转换反应中通过应变工程、电子效应等策略提高催化性能。总结已有合成方法,直接液相合成方法是制备大规模、小尺寸、高度分散的铂基金属间化合物的最有潜力的方法。Pt基金属间化合物纳米晶的发展潜力巨大,将来很有可能在以下几方面进行突破。1)在制备研究方面:通过相图可以筛选出可以制备Pt基金属间化合物纳米晶催化剂的元素,为实验提供指导。然而,由于块体材料与纳米材料由于尺寸带来的差异性,即使筛选出的Pt基金属间化合物在热力学(相图)上是稳定的,但在实际合成反应中仍存在挑战。此外,开发出普适性强、低成本的大规模可控制备技术是Pt基金属间化合物催化剂迈向商业化的必经之路。2)催化表征方面:借助原位谱学技术和理论计算可以深入理解各类电催化过程中界面处的电荷转移,从而建立优化催化剂的理论指导。3)理论计算:由于金属间化合物家族庞大,仅仅依靠实验筛选,将浪费大量人力物力,未来可以借助大数据分析、机器学习和理论模拟等手段对催化剂进行快速筛选来指导实验,目前来看机器学习指导DFT计算的方法有很大潜力,但仍处在探索阶段。相信随着未来对铂基金属间化合物纳米晶催化机理的深入认识及精准可控合成技术的快速发展,金属间化合物催化剂将在燃料电池等能源转化装置中实现商业化应用。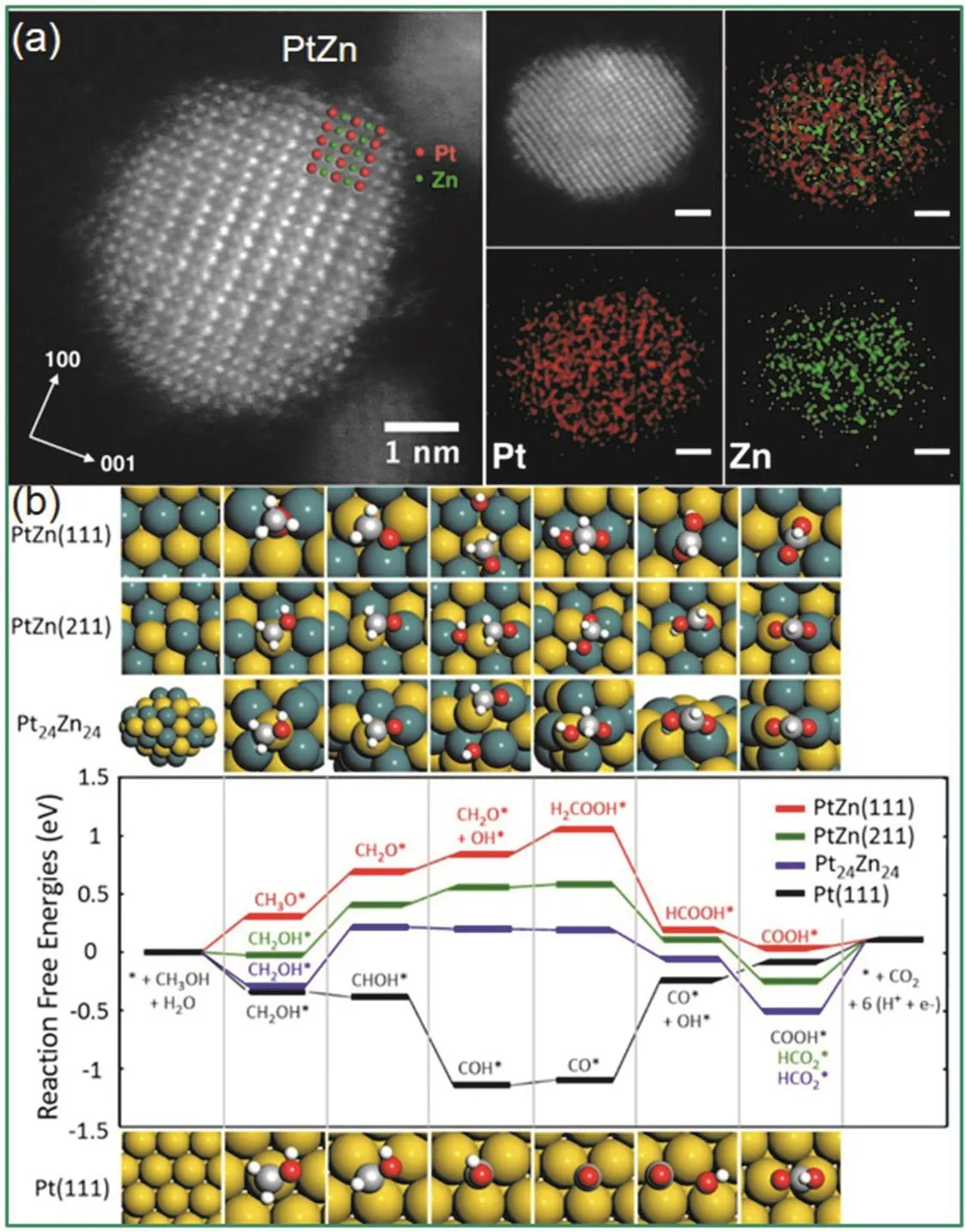
3.3 理论计算助力电催化剂筛选
4 总结与展望
