基于环糊精本征识别能力的手性色谱介质点击制备及应用
2020-09-23靳晓宁马骁飞
陈 明, 靳晓宁, 马骁飞, 王 勇
(天津大学理学院, 天津 300350)
环糊精[1](cyclodextrin, CD)是一类环状寡聚糖,由呈椅式构象的D-吡喃葡萄糖单元以1,4-糖苷键[2,3]结合成环,由于连接葡萄糖单元的糖苷键不能自由旋转,所以CD分子不是圆筒状而是略呈锥形的中空圆筒立体环状结构,在其空洞结构中,较大开口端由C-2和C-3的仲羟基构成,较小开口端由C-6的伯羟基构成,CD最显著的特征是其亲脂性空腔可与众多非极性分子形成主客体包合物[4]。衍生后的CD除了具有CD本身的主客体包合作用外,还具有π-π相互作用、偶极-偶极相互作用、空间位阻效应等多重作用力,在手性分离领域有非常广泛的应用。
高效液相色谱[5,6](HPLC)是目前最常用的分离和纯化技术之一。CD及其衍生物被广泛用作色谱中的手性选择剂,通过将CD固载在硅胶上可获得色谱中的手性固定相(CSP)。CD固载在硅胶上主要有两种方法,一种是物理涂覆,另一种是化学键合。物理涂覆可以直接将具有官能团的CD涂覆于硅胶上从而获得新型的CSP。化学键合是通过强大且稳定的键合臂作用将硅胶和CD结合起来,被认为是一种更好的方法用于制备CD-CSP。自1983年Fujimura首次成功地基于氨基合成CD-CSP[7]以来,它们在分离领域引起了极大的兴趣。化学家们一直在寻找制备稳定CD-CSPs的合成方法,具有各种键的CD-CSPs得到广泛开发,例如Armstrong开发的醚键CD-CSPs[8,9], Ng发展的脲基键合臂CD-CSPs[10]等。但是传统的制备方式存在一系列的问题,如易水解、结构不明确、制备重现性低及条件苛刻[11]等,因此最近的大多数研究都集中在结构明确的CD-CSPs的开发,其方法是寻求有效温和的化学键合方法和CD羟基的功能化以提高对映选择性。Cu(Ⅰ)催化的1,3-偶极环加成反应[12]的出现成为制备稳定CD-CSPs的重要手段。2008年,Liang和Ng两个研究组分别利用该反应合成了三唑键合臂的天然CD-CSP,并成功应用于HPLC中,对多种药物实现了分离[13,14]。Ng课题组采用此反应合成的苯基异氰酸酯基衍生化CD-CSP实现了对6组碱性和中性对映体的快速拆分[15]。之后,许多课题组对该反应进行了深入的研究,并成功制备了衍生化的CD-CSPs,引入了其他功能基团[16,17]。随后,“巯基-烯”的点击反应也得到了广泛应用[18,19]。我们课题组在此基础上合成了结构明确且性质非常稳定的CD-CSPs[20,21],并对异唑啉类、黄烷酮类等多种手性药品实现了有效拆分。
上述研究大多集中于对CD或桥联臂进行功能衍生引入更多作用位点以提升手性拆分能力,但目前鲜有能够反映天然CD本征识别能力的手性固定相的研究报道,是否引入官能团有利于哪些类手性对映体的拆分并不十分清楚,如何充分发挥环糊精本征的手性识别作用还有待进一步研究。本文通过“巯基-烯”点击化学反应合成了能够反映环糊精本征手性拆分能力的单(6-巯基-6-去氧)-β-环糊精手性固定相(CSP1),考察了其对50多种手性药物的手性识别能力,包括异唑啉、手性交酯、手性酮、黄烷酮以及丹磺酰(Dns)氨基酸等,并进一步与功能三唑桥联CD-CSP(CSP2)及咪唑嗡桥联CD-CSP(CSP3)在同一色谱条件下进行了对比,为后期CD色谱固定相结构的设计提供参考。
1 实验部分
1.1 仪器、试剂与材料
LCQ Deca XP MAX液相色谱-质谱联用仪(美国Thermo Fisher), Infinityplus 300固体核磁共振谱仪(美国瓦里安公司), FTS3000傅里叶红外光谱仪(天美科学仪器有限公司), Vario Micro cube元素分析仪(德国Elementar), PHS-2F pH酸度计(上海雷磁仪器厂), P10SNXP1高压填柱泵(美国LabAlliance)。
偶氮二异丁腈(AIBN)、偏重亚硫酸钠(Na2S2O5)、无水N,N-二甲基甲酰胺(DMF)和乙烯基三乙氧基硅烷均购自天津希恩思生化科技有限公司(天津),丙酮、无水乙醚和甲苯均购自利安隆博华医药化学有限公司(天津),硫脲、三氯乙烯和无水甲醇购自天津市元立化工有限公司(天津),硅胶(5 μm, 10 nm)购自Fuji Silysia (Fuji,日本), HPLC级甲醇和三乙胺(TEA)购自天津市康科德科技有限公司(天津)。根据文献[22]的方法合成了单-6-对甲苯磺酰基-β-CD (TsO-CD)。实验中用作手性分离的外消旋体的结构如图1所示,异唑啉类样品参照文献[23]报道合成,手性交酯[24]和手性酮类[25]样品由中国科学院福建物质结构研究所康强教授提供,丹磺酰氨基酸类样品购自Sigma-Aldrich公司(上海),黄烷酮类样品购自上海安耐吉化学公司(上海)。原装液相色谱柱(150 mm×4.6 mm)即不锈钢柱购自Daicel公司(日本)。
1.2 仪器和方法
采用Hitachi液相色谱分析仪进行色谱分析,配有二极管阵列检测器(DAD),检测波长为254 nm。流动相为MeOH/H2O(1∶1, v/v)和MeOH/三乙胺-乙酸缓冲液(1% (v/v) 三乙胺,乙酸调节pH至5.30)(1∶1, v/v),流速为0.5 mL/min。每个样品均重复进样3次,所得结果取平均值。保留因子(k)、选择因子(α)和分离度(Rs)均采用美国药典(USP)标准进行计算,所用公式为:k=(tR-t0)/t0(tR为对映体的保留时间,t0为死时间,由基线波动所决定);α=k2/k1;Rs=1.18×(t2-t1)/(Wh1+Wh2)(Wh是色谱峰的半峰宽值)。
1.3 CSP1的合成
按图2所示思路采用“巯基-烯”点击反应进行。
1.3.1单(6-巯基-6-去氧)-β-CD的合成[26]
将TsO-CD(3.00 g, 2.33 mmol)和硫脲(1.77 g, 23.3 mmol)溶解在DMF(120 mL)中,在75 ℃下搅拌48 h。在室温下冷却后,将混合物加入500 mL乙醚中并搅拌10 min。过滤沉淀物并用100 mL乙醚洗涤,之后悬浮在120 mL丙酮中,并加热回流2 h。将悬浮液冷却至室温并过滤,收集白色固体真空干燥后分批加入含有120 mg Na2S2O5的1 mol/L NaOH(100 mL)溶液中,搅拌30 min后,用浓HCl将溶液酸化至pH=3。将5.3 mL三氯乙烯加入到反应混合物中,并将所得悬浮液超声处理10 min。过滤收集白色固体,真空干燥,得到目标产物1.83 g。
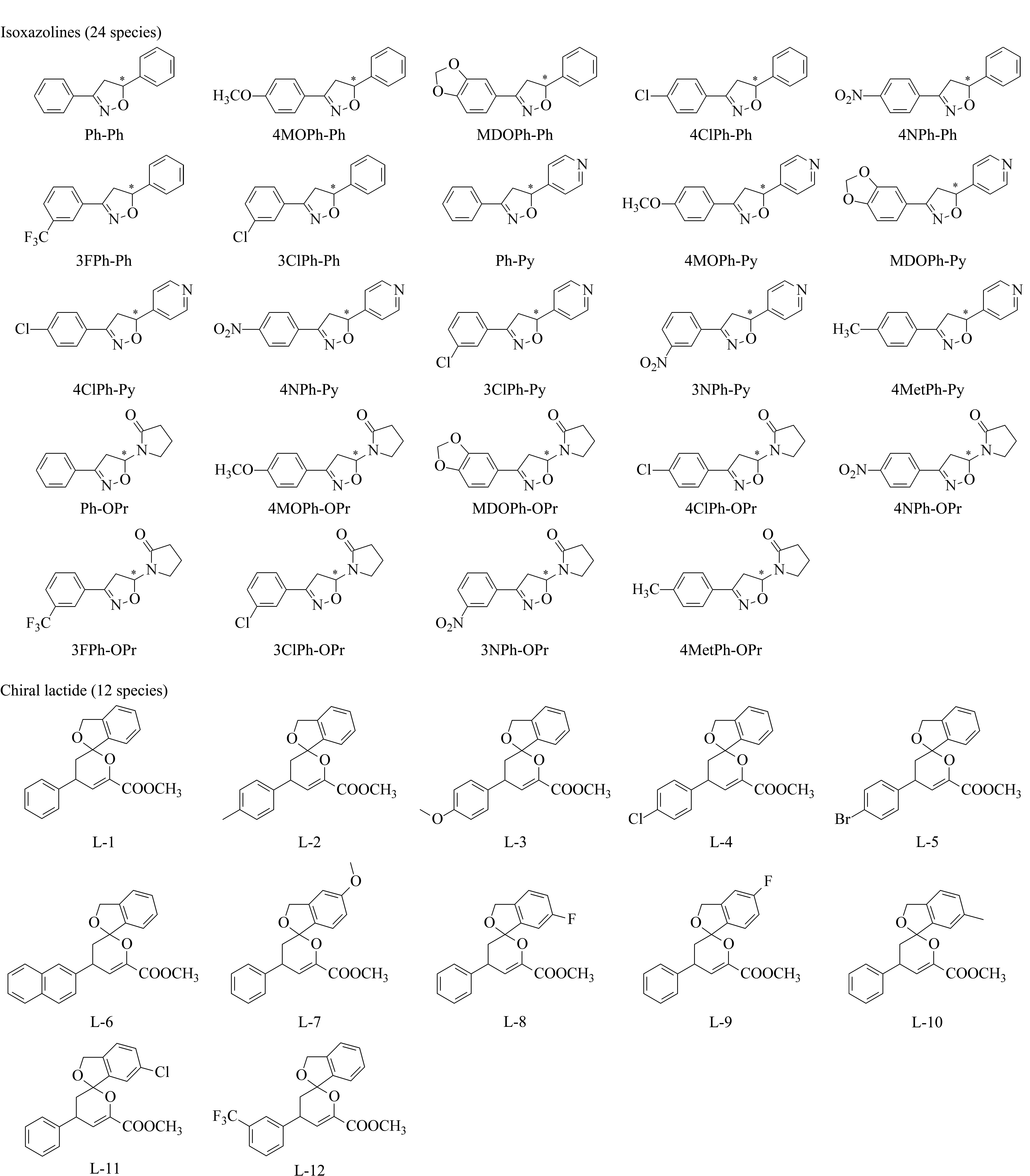
图 1 消旋体的结构式Fig. 1 Structures of the racemates

图 1 (续)Fig. 1 (Continued)
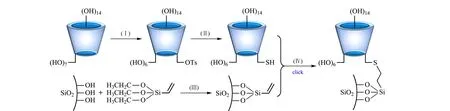
图 2 CSP1的合成路线Fig. 2 Synthetic route of CSP1 Ⅰ: synthesized according to reported methods[22]. Ⅱ: synthesized according to reported methods[26]. Ⅲ: anhydrous toluene, heated at 120 ℃ under reflux for 24 h. Ⅳ: mixed solution of methanol and deionized water (1∶1, v/v), azodiisobutyronitrile (AIBN) catalyzed, stirred at 60 ℃ for 24 h. Ts: toluene sulfonyl.
液体核磁共振(1H-NMR)(400 MHz, DMSO-d6)δ: 2.1 (t, SH), 2.7~3.2 (m, 2H, H-6a), 3.2~3.5 (m, 与HDO峰重叠, H-2, H-4), 3.5~3.8(m, 26H, H-3, H-5, H-6b), 5.80~5.61(m, 14H, OH-2, 3), 4.87~4.83(m, 7H, H-1), 4.55~4.47(m, 6H, OH-6)。
红外光谱(FTIR)(cm-1, KBr): 3 388(O-H伸缩振动), 2 923(C-H伸缩振动), 2 566(弱吸收峰,SH的伸缩振动), 1 651(O-H弯曲振动), 1 030(C-O-C对称伸缩振动)。
电喷雾电离质谱(ESI-MS)(m/z):计算值(C42H70O34S)1 151.05 [M+],实测值1 173.34 [M+Na]+。
1.3.2双键功能化硅胶的合成
将3.0 g活化硅胶(120 ℃真空干燥过夜)均匀分散到30 mL无水甲苯中,向反应溶液中加入乙烯基三乙氧基硅烷1.3 mL, N2保护下,在120 ℃加热回流24 h,之后过滤得固体粗产物,将粗产物用丙酮索氏提取24 h,真空60 ℃干燥得到目标产物2.74 g。
FTIR(cm-1, KBr): 3 443(O-H伸缩振动), 2 985(=C-H伸缩振动), 2 908(C-H伸缩振动), 1 658(O-H弯曲振动), 1 068(C-O-C对称伸缩振动)。
1.3.3“巯基-烯”点击反应制备CSP1
向甲醇和去离子水(1∶1, v/v)的混合溶液(40 mL)中加入单(6-巯基-6-去氧)-β-CD 1.5 g,充分搅拌使其完全溶解。之后向反应体系中加入3 g双键硅胶和50 mg催化剂AIBN。N2保护下,在60 ℃下搅拌反应24 h。反应完成后,将反应体系过滤得到粗产物,依次用蒸馏水(2×20 mL)、甲醇(2×20 mL)、丙酮(2×20 mL)洗涤,真空60 ℃干燥得到目标产物3.66 g。

表 1 双键功能化硅胶和CSP1的元素分析
1.4 参比手性色谱介质的合成
根据参考文献[27,28]报道的方法分别合成了功能三唑桥联CD-CSP(CSP2)及咪唑嗡桥联CD-CSP(CSP3)作为参比手性介质进行实验对照(CSP2和CSP3的表面CD固载量分别为0.51 μmol/m2, 0.46 μmol/m2)。
1.5 色谱柱的填装
采用高压匀浆法将所制备的手性介质装入不锈钢柱(150 mm×4.6 mm)中,甲醇作为分散剂,装柱压力为41.37 MPa,装柱时间为30 min左右。在使用之前将色谱柱用甲醇冲洗,并采用流动相进行平衡。
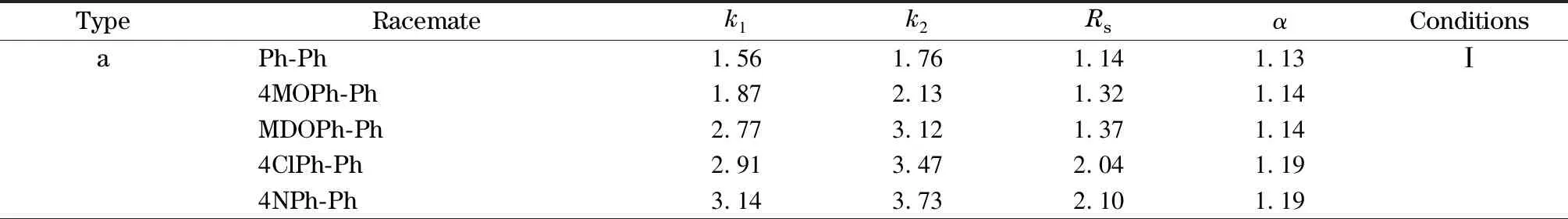
表 2 样品在CSP1上的分离
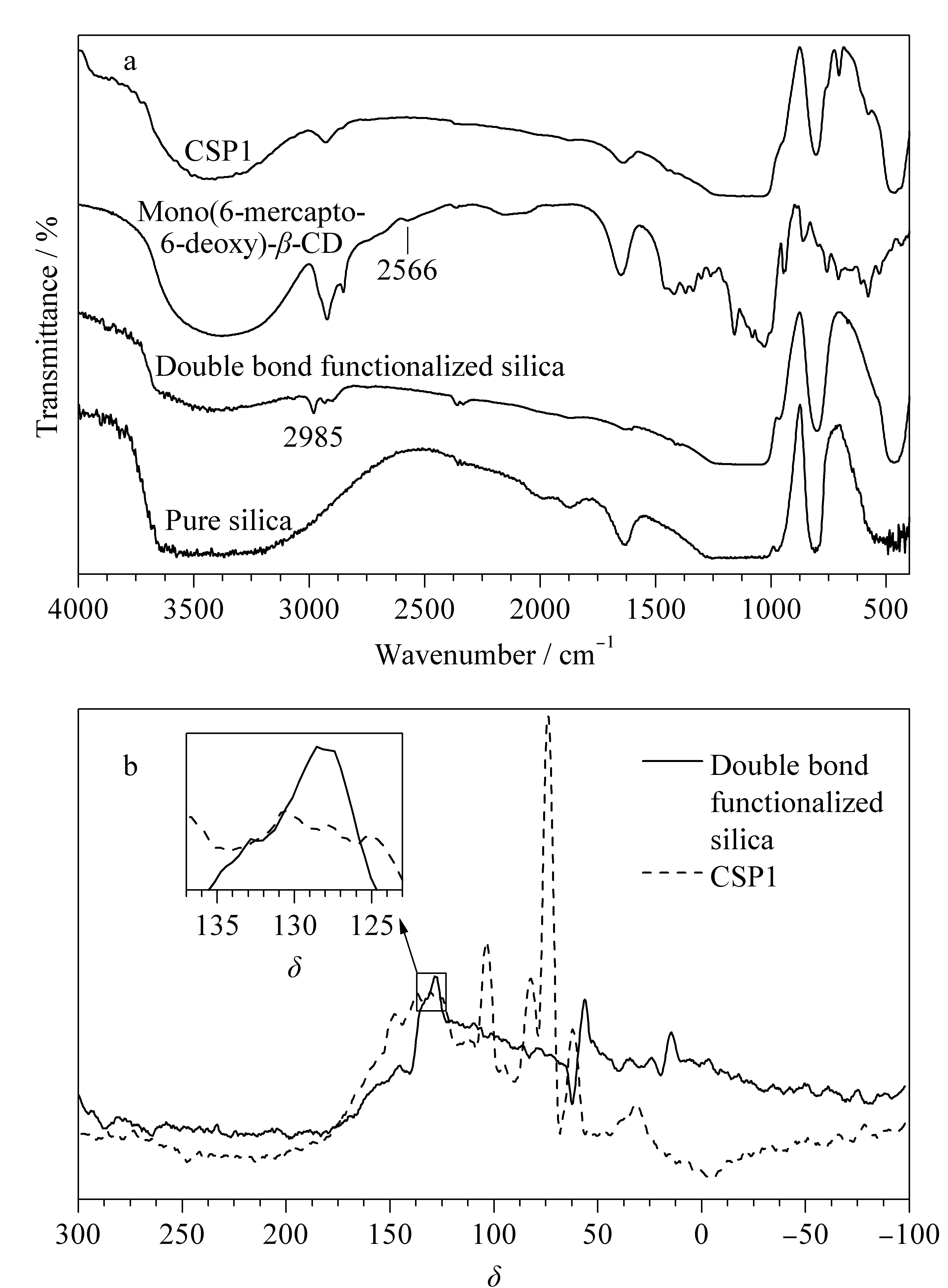
图 3 产物的(a)FTIR和(b)13C SSNMR表征对比图Fig. 3 Product characterization comparison chart (a) FTIR, (b) 13C SSNMR
2 结果与讨论
2.1 CSP1的表征
为了证明CSP1可以通过“巯基-烯”点击反应合成,采用FTIR、固体核磁共振(13C SSNMR)和元素分析对产物进行了表征。在FTIR光谱(见图3a)中,相比于裸硅胶,双键功能化硅胶在2 985 cm-1处出现了=C-H伸缩振动峰,这说明双键已经成功地键合至硅胶表面。另外,单-6-SH-β-CD在2 566 cm-1处有一个弱吸收峰,此峰为-SH的伸缩振动峰。在进行“巯基-烯”点击反应后,在2 930 cm-1附近出现了CD亚甲基的吸收峰,且=C-H和-SH的特征吸收峰均消失。13C SSNMR(图3b)中,双键功能化硅胶在160~90的双键碳原子信号峰在合成CSP1后消失,在110~65处出现了新的CD碳原子信号峰。13C NMR和FTIR结果表明成功制备了CSP1。元素分析结果(见表1)表明,与双键功能化硅胶相比,CSP1的C、H、N的百分含量均得到了提高,计算得出CSP1表面CD固载量为0.82 μmol/m2。
2.2 CSP1的手性识别能力
为了考察CSP1的本征手性识别能力,本文选取了5大类包括异唑啉、手性交酯、手性酮类、黄烷酮类,丹磺酰氨基酸等50多种消旋体用于评价该手性固定相的拆分性能,结果见表2。

表 2 (续)
2.2.1异唑啉类样品的分离
由表2a可知,Ph-Ph类样品在CSP1上表现出较好的分离效果,其Rs值均大于1,这主要利用的是环糊精本征的主客体包合作用,该作用与分子间氢键、空间位阻效应等相结合,可以形成有效的手性拆分三点作用模型。其中,4ClPh-Ph和4NPh-Ph在CSP1上Rs的值均大于1.5,达到了基线分离,4ClPh-Ph是由于-Cl基团的吸电子效应,4NPh-Ph是由-NO2与CD形成的氢键作用导致的。另外,4ClPh-Ph的Rs值均大于3ClPh-Ph的Rs值,这表明取代基的位置不同会导致不同的包合作用和分离结果,客体分子苯环上的3位取代不利于CSP1手性识别。综上所述,环糊精的本征识别能力比较有利于含有两个疏水苯环基团Ph-Ph类样品的分离。
由表2b可知,Ph-Py类样品在CSP1上的分离效果较差,仅4NPh-Py和4MetPh-Py表现出分离迹象,然而其Rs值小于0.8,并未达到基本分离,这说明吡啶环对样品的分离有一定的抑制作用,由于环糊精外侧羟基可与吡啶环氮原子形成氢键,阻碍了包合作用的进行,进而导致该类对映体分离效果较差。
由表2c可知,除3FPh-OPr外,其他Ph-OPr类样品在CSP1上均获得了部分分离,但效果并不理想,未达到基线分离,这说明样品分子中吡咯烷酮基团虽然也可与环糊精形成较好的包合,但由于极性较大,与苯环相比包合作用较弱,无法支撑较好的手性分离结果。其中,MDOPh-OPr、4ClPh-OPr、4NPh-OP和4MetPh-OPr在CSP1上达到了较好的分离,这主要是由于这些样品所连基团提供的相应作用力提高了分离效果。另外,通过比较4ClPh-OPr和3ClPh-OPr以及4NPh-OPr和3NPh-OPr的Rs值可知,苯环上4位取代的化合物分离效果均好于3位取代的化合物,这同样说明苯环上的3位取代不利于CSP1的手性识别。
2.2.2手性交酯类样品的分离
手性交酯类样品的分离结果如表2d所示。根据该类样品的分子结构特点,可以将这12种样品分为3类,第一类为L-1,该样品的六元环和苯并呋喃环上无任何取代基,其在CSP1上仅仅表现出分离迹象,但未达到基线分离。第二类为L-2~L-6, L-12,该类样品的主要特点是六元环上取代基的种类和位置不同,其中,L-2~L-6为4位取代,L-12为3位取代,分析可知,其分离效果相差无几,这充分说明六元环上取代基的位置对此类样品的分离效果影响不大。对于L-2~L-6的分离,仅L-6的Rs值大于1.5,这是由于萘基能够更好地契合环糊精空腔大小,起了主导作用;第三类为L-7~L-11,该类样品的主要结构特点是苯并呋喃环所连的取代基不同,该类样品在CSP1上的分离效果并不是很好,均只表现出分离迹象,但仅有L-8和L-10达到了基线分离,L-8是由于-F提供的氢键作用,L-10是由于-CH3的供电子作用;另外,L-8和L-9均连有-F,但L-8的分离效果比L-9好,这说明苯并呋喃环上取代基的位置不同会对分离效果造成影响。
总之,在手性交酯类样品的分离中,这3类样品的分离效果均不是很好,环糊精的本征识别能力适用于部分手性交酯样品的分离。
2.2.3手性酮类样品的分离
由表2e分析可知,手性酮类样品在CSP1上均获得一定分离,个别样品达到了较好的分离效果。其中,D-6的Rs值达到了5.46,这是由于环糊精的空腔能够更好地匹配D-6分子结构以及-Cl的吸电子效应起到了主导作用;D-4和D-5的Rs值均大于1,达到了基本分离,这是由于-CH3的供电子作用和苯环的共轭效应,但可能由于-CH3和苯环的空间位阻效应,导致其分离效果不如D-6; D-1~D-3仅表现出分离迹象,其Rs值均小于0.8,这说明环糊精的本征空腔不利于这3个样品的分离。
综上所述,在手性酮类样品的分离中,样品较大的空间位阻效应抑制了环糊精的本征识别能力,因此此类样品在CSP1上的分离效果并不理想,需要借助外在功能团进一步调控其手性识别性能。
2.2.4丹磺酰氨基酸类样品的分离
由表2f可知,大部分丹磺酰氨基酸类样品在CSP1上均实现了较好的分离,这是由于CD空腔和萘基部分形成了良好的包合作用、静电作用,并且与手性碳相邻的脂肪族侧链引起的空间效应有助于手性选择性的提高。其中,Dns-DL-Leu、Dns-DL-Aca和Dns-DL-Apa均取得了非常好的分离效果。这除了与环糊精的本征识别能力有关外,还与其自身所连的基团有关,Dns-DL-Leu是由于两个甲基的空间位阻作用,Dns-DL-Aca是由于其脂肪族侧链的疏水作用,Dns-DL-Apa是由于-OH的氢键作用和空间位阻效应;Dns-DL-Val、Dns-DL-Phe、Dns-DL-Nle和Dns-DL-Thr的Rs值大于1,达到了基本分离,这说明环糊精的本征识别能力有利于这4个样品的分离,但其所连的基团起到了一定的抑制作用;另外,有如下规律:Rs(Dns-DL-Aca)>Rs(Dns-DL-Nle)>Rs(Dns-DL-Nva)>Rs(Dns-DL-ABa),且Rs(Dns-DL-Leu)>Rs(Dns-DL-Val),这充分说明,脂肪族侧链疏水作用越强,其分离效果越好。
总之,除了环糊精本征识别能力产生的影响外,丹磺酰氨基酸的分子结构也会对其分离效果产生重要影响,此外,取代基中的侧链数量同样对分离效果的好坏产生一定的影响,加长其侧链或提高疏水性可有效提升分离效率。
2.2.5黄烷酮类样品的分离
由表2g可知,黄烷酮类样品在CSP1上的分离效果一般。其中,6-甲氧基黄烷酮和7-甲氧基黄烷酮的分离效果较为可观,且7-甲氧基黄烷酮的Rs值大于1,这主要是-OCH3供电子作用力的影响,且其取代基的位置不同也对分离效果造成了影响。
2.2.6部分样品在CSP1中分离的典型色谱图
部分样品在CSP1中分离的典型色谱图见图4。
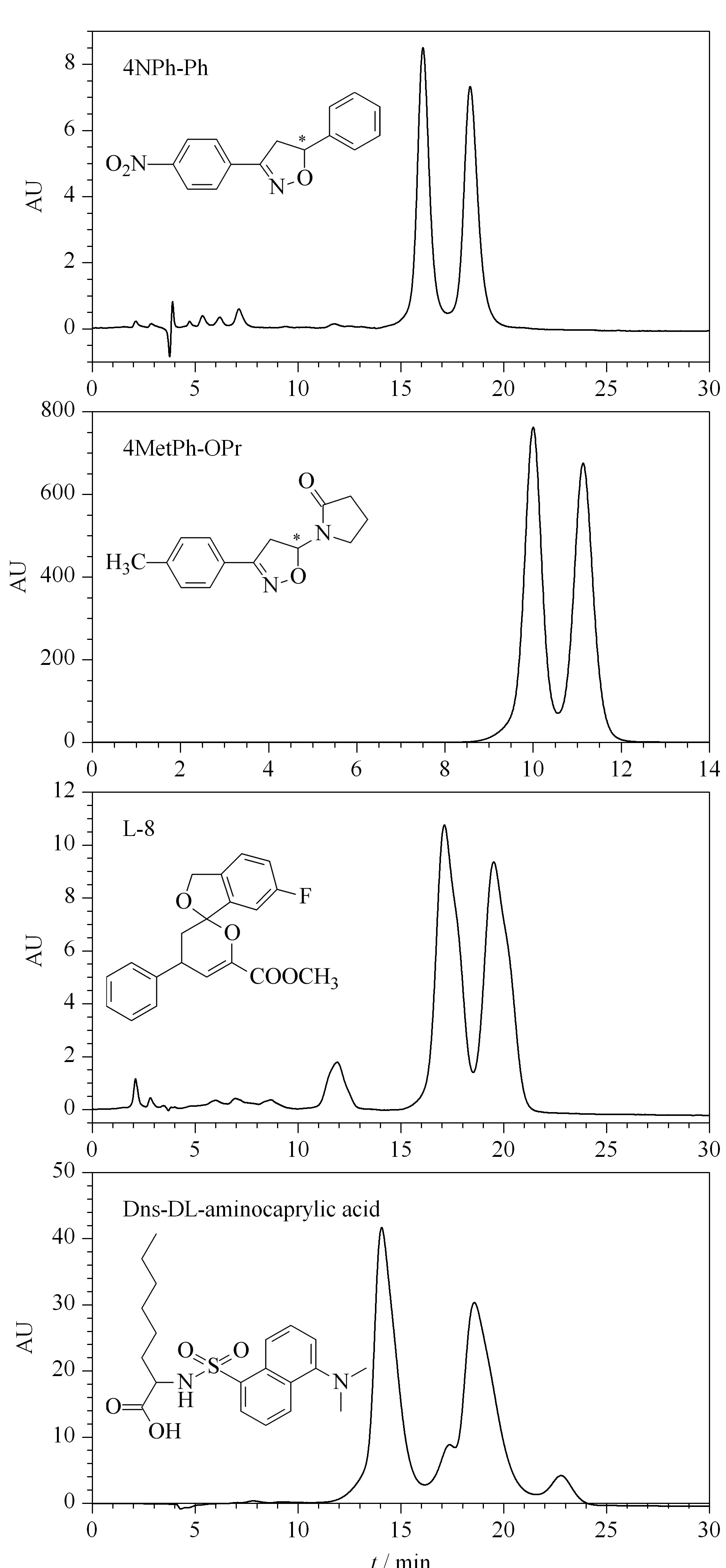
图 4 样品在CSP1上分离的典型色谱图Fig. 4 Typical chromatogram of sample separation on CSP1
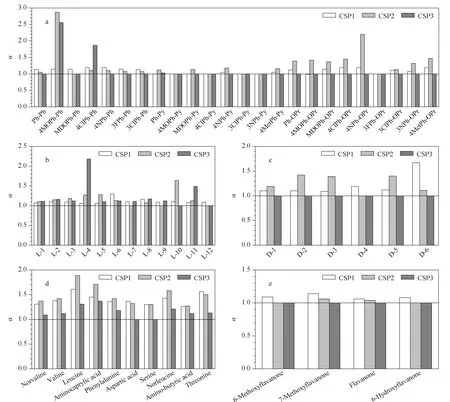
图 5 消旋体在CSP1~3上的手性分离效果对比图Fig. 5 Comparison of chiral separation effects of racemates on CSP1-3a. isoxazolines; b. chiral lactides; c. chiral ketones; d. dansyl amino acids; e. flavanones.
2.3 CSP1与CSP2、CSP3手性识别能力的对比
为进一步探究环糊精的本征识别能力,将CSP2和CSP3的手性色谱柱与CSP1在同一色谱条件下进行了实验对照,具体分离效果见图5。由图5a可知,在异唑啉3类样品的分离中,Ph-Ph类样品在CSP1上的分离效果最好;Ph-Py类样品CSP1~3上的分离效果均较差,相比之下,在CSP2上的分离效果最好;Ph-OPr类在CSP3上均未分离,在CSP1和CSP2中,除3FPh-OPr外,其他样品均达到了一定程度的分离。由图5b可知,六元环和苯并呋喃环上无任何取代基的L-1在CSP1~3上都表现出分离迹象,但均未达到基本分离;六元环上含取代基的L-2~L-6, L-12, CSP2的分离效果最好,其次是CSP1,分离效果最差的是CSP3;苯并呋喃环连有取代基的L-7~L-11在CSP1~3上的分离效果均不是很好。由图5c可知,手性酮类样品在CSP2上的分离效果最好,其次是CSP1,而在CSP3上均未出现分离迹象。由图5d可知,除个别样品外,丹磺酰氨基酸类样品在CSP1~3上均实现了一定程度的分离,相比之下,此类样品在CSP2上的分离效果最好,在CSP3上的分离效果最差。由图5e可知,黄烷酮类样品在CSP1~3上的分离效果均不理想,其在CSP1上的分离效果最好,但也未达到基本分离。总之,环糊精的本征识别能力仅利于部分样品的分离,并不适用于所有样品,对桥联臂进行功能改性虽然能够提升其对部分样品的拆分能力,同时也会小幅损失CD色谱固定相的手性识别能力。
3 结论
本文通过“巯基-烯”点击化学反应合成了桥联臂无官能团的单(6-巯基-6-去氧)-β-环糊精手性固定相 ,其最大限度地保留了环糊精的本征结构,采用HPLC反相模式探究了该手性介质对50多种消旋体的识别能力,并进一步将其与功能三唑桥联CD-CSP及咪唑嗡桥联CD-CSP在同一色谱条件下进行了结果比对。通过比较可知,样品的分离过程除了与手性介质的结构有关外,还与样品分子的结构有很大关系,对桥联臂进行功能改性可提升对部分对映体的选择性,但同时会小幅损失CD的本征手性识别能力。对于环糊精本征识别能力易于分离的样品,在设计手性介质时,其桥联臂不需要任何官能团,探讨环糊精的本征识别能力也为手性介质的结构设计提供了有益参考。
