超支化聚酰胺改性海藻酸钠微球对Cr(Ⅵ)的吸附
2020-09-17王莉莉余德游王懿佳吴明华
李 恒,王莉莉,,余德游,王懿佳,吴明华,3,王 炜
(1.浙江理工大学生态染整技术教育部工程中心,浙江杭州 310018;2.三元控股集团有限公司,浙江杭州 311221;3.浙江理工大学先进纺织材料与制备技术教育部重点实验室,浙江杭州 310018)
在印染加工中,酸性媒介染料染色时使用媒染剂重铬酸钾、感光制网时使用重铬酸盐型光敏剂以及含铬络合染料,造成大量铬进入水体,引起印染废水含铬量超标。铬是毒性最强的重金属之一,主要以Cr(Ⅲ)和Cr(Ⅵ)的形式存在于水体中。Cr(Ⅵ)具有相当大的毒性,其毒性大约是Cr(Ⅲ)的500 倍,在自然环境中难以生化降解,且可以通过食物链在人体内富集,从而引发严重的健康问题,如致基因突变、致癌、致肝肾损伤等[1]。因此,如何有效地处理含Cr(Ⅵ)的废水,是当今环保领域的重要研究课题。吸附法因适应性好、可回收、可重复利用、效率高、操作简单,是业内公认综合效能较佳的重金属处理方法[2],吸附剂是决定高效能吸附的关键因素。因此,开发价廉、高性能、可再生的环境友好型吸附剂是重金属污染控制与治理的关键,也是重金属吸附研究的发展方向。
近年来,研究人员从环境保护角度考量,更多地关注天然可再生生物质材料,以此去除废水中的重金属离子[3]。海藻酸钠(SA)是用于去除重金属离子的众多天然生物质材料之一,具有来源广、无毒、可生物降解等优点。海藻酸钠是由β-D-甘露糖醛酸(M)和α-L-古洛糖醛酸(G)组成的线性阴离子水溶性多糖,分子骨架上含有丰富的羟基(—OH)和羧酸根离子(—COO-),提供了与多种多价金属离子螯合的位点。粉末状海藻酸钠直接用于去除重金属离子不便于分离和回收再利用,因此,一般将海藻酸钠用不同的交联剂制成多孔膜或凝胶球后用于吸附重金属离子[4-5]。而交联剂的使用会占据海藻酸钠的功能性基团,影响其对重金属离子的吸附能力。Karthik等[6]研究了Ca2+交联的海藻酸钠珠粒去除水溶液中Cr(Ⅵ)的能力,最大吸附量仅为24.2 mg/g。因此,进一步探索具有高吸附量和环境友好的吸附剂对于实际应用具有重要意义。
目前的研究主要集中在对SA 进行功能基团改性后再交联上,该方法在一定程度上改善了SA 的吸附性能,但仍然存在一定的不足。在交联改性过程中,吸附功能基团仍有一部分参与了交联,并且大多数吸附功能基团被包埋在微球内部,只有部分功能基团裸露在微球表面发挥吸附作用。为了克服上述缺陷,本研究采取先制备微球再接枝功能基团的策略[7],即先制备SA 微球,再接枝超支化聚酰胺(HA),有效增加Cr(Ⅵ)的吸附位点。胺基在酸性条件下易质子化,可通过静电作用和离子交换来去除Cr(Ⅵ),而超支化聚酰胺具有三维结构和丰富的亚氨基、伯氨基[8-9]。将超支化聚酰胺接枝到SA 微球表面,引入大量酰胺基和氨基功能基团,可以改善SA 的吸附性能,同时功能基团最大程度地暴露在SA 微球表面,增强了对Cr(Ⅵ)的吸附效果。此外,胺基功能化SA 微球具有球状结构,避免了粉末吸附材料分离回收难的问题。目前,关于超支化聚酰胺改性SA 微球的研究尚没有报道。
本实验以戊二醛作为交联剂,将自制的HA 接枝到SA 微球表面,制备出超支化聚酰胺改性海藻酸钠(HA@SA)微球吸附剂,并应用于去除印染废水中的Cr(Ⅵ)。通过FT-IR、EDS、XPS 等表征HA@SA 微球吸附前后结构的变化。通过批次吸附实验考察溶液pH、Cr(Ⅵ)初始质量浓度、吸附时间以及吸附温度等对HA@SA 微球吸附性能的影响。通过研究吸附动力学、吸附等温线和吸附热力学,探讨HA@SA 微球的吸附机理。通过吸附-解吸实验探讨HA@SA 微球的循环再生能力。
1 实验
1.1 材料与仪器
试剂:海藻酸钠(SA,化学纯,山东洁晶集团股份有限公司),氯化钙(CaCl2)、乙醇(C2H5OH)、氢氧化钠(NaOH)、硝酸(HNO3)(分析纯,杭州高晶精细化工有限公司),戊二醛(GA,分析纯,上海麦克林生化科技有限公司),盐酸(HCl,分析纯,杭州双林化工试剂有限公司),重铬酸钾(K2Cr2O7,分析纯,天津市科密欧化学试剂有限公司),超支化聚酰胺(HA,自制)。
仪器:CP114 型电子天平[奥豪斯仪器(上海)有限公司],DF-101S型集热式恒温加热磁力搅拌器(上海棱标仪器有限公司),AD2.0E 型冷冻真空干燥仪(美国VIRTIS 公司),SHA-B 型水浴恒温振荡器(常州亚特实验仪器有限公司),AA-110 型原子分光光度计(美国瓦里安公司),FE28 型pH 计(上海梅特勒-托利多仪器有限公司),Nicolet 5700 型傅里叶红外光谱仪(FT-IR,美国热电公司),Brookhaven Zeta PLAS高分辨Zeta 电位仪(美国布鲁克海文仪器公司),KAlpha 型X 射线光电子能谱仪(XPS,美国赛默飞世尔科技公司),EDS649 型能量色散谱仪(EDS,英国牛津仪器分析有限公司)。
1.2 HA@SA 微球的制备
将2.0 g SA 溶于100 mL 去离子水中,搅拌溶解成黏稠液体;30 min 内将SA 溶液逐滴加入100 mL 1%的CaCl2水溶液中,室温下搅拌6 h,过滤得SA 微球;用去离子水洗涤微球多次以除去微球表面杂质。将SA 微球放入100 mL 2%的戊二醛溶液中,加入2 mL 1 mol/L 的HCl 溶液作为催化剂,搅拌下60 ℃交联反应6 h,过滤,用去离子水洗涤微球多次。将微球加入100 mL 20 g/L 的HA 溶液中,在搅拌下50 ℃接枝反应1 h[8],过滤,用去离子水洗涤微球数次后,用乙醇洗去微球表面未反应的HA,得到HA@SA 微球。
1.3 批次吸附实验
在一定温度、pH 条件下,将0.027 g HA@SA 微球加入10 mL Cr(Ⅵ)溶液中,振荡吸附一定时间,Cr(Ⅵ)质量浓度采用火焰原子吸收分光光度法(FAAS)测定[10](用0.1 mol/L HCl 或NaOH 调节pH)。平行测定3次,取平均值。平衡吸附量(qe)计算公式如下:

其中,ρ0和ρe分别是Cr(Ⅵ)的初始质量浓度和平衡质量浓度,mg/L;V是被吸附溶液的体积,L;m是HA@SA微球(干燥)质量,g。
吸附动力学实验在400 mg/L 的Cr(Ⅵ)溶液中进行,在不同时间点取样,测定Cr(Ⅵ)的质量浓度;吸附等温线实验在溶液pH 为2.0 的条件下进行,吸附26 h,Cr(Ⅵ)初始质量浓度为100~1 300 mg/L;吸附热力学实验在30~65 ℃400 mg/L 的Cr(Ⅵ)溶液中进行。
1.4 吸附-解吸再生实验
在30 ℃条件下,将0.027 g HA@SA 微球加入10 mL 400 mg/L 的Cr(Ⅵ)溶液中,调节pH 为2.0,振荡吸附26 h,过滤,测试残液的Cr(Ⅵ)质量浓度。用去离子水清洗负载Cr(Ⅵ)的HA@SA 微球后转移到50 mL 含0.1 mol/L NaOH 和0.1 mol/L NaCl 的溶液中进行解吸实验[8](在室温下振荡解吸1 h),过滤后用蒸馏水洗涤至中性。循环重复以上过程8次。
保留率是吸附剂第i次循环再生后的吸附量(qi,mg/g)与第1次吸附量(q1,mg/g)的比值:
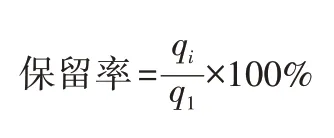
保留率越高,吸附剂第i次循环再生后的吸附量越大,吸附剂的再生性越好。
1.5 测试
FT-IR:在400~4 000 cm-1采用KBr 压片法测试;EDS:采用能谱仪进行元素分析;XPS:采用X 射线光电子能谱仪[配备有Al-Kα X 射线源(1 486.6 eV)]通过压片法测试。
2 结果与讨论
2.1 表征
2.1.1 FT-IR
对比图1a,图1b 中,O—H 伸缩振动峰从原来的3 382 cm-1移动到3 425 cm-1,并且峰强度显著增加,这是由接枝超支化聚酰胺后胺基的氢键引起;此外出现了2 927、2 852 cm-1新峰,归因于超支化聚酰胺结构上—CH 和—CH2的C—H 伸缩振动;1 612 cm-1处出现CN 特征吸收峰,表明HA 上的胺基与羰基反应生成了席夫碱。这些变化表明HA 已经成功接枝到SA 表面。图1c 中,Cr(Ⅵ)负载到HA@SA 微球上的特征吸收峰为802 cm-1[8];此外,O—H 和N—H 吸收峰从3 425 cm-1偏移到3 322 cm-1,这是由吸附剂和铬离子之间的静电作用导致,证实Cr(Ⅵ)已经吸附到HA@SA 微球表面。
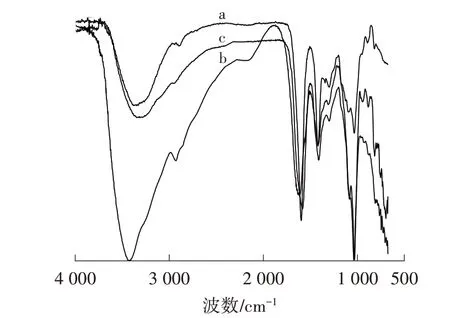
图1 SA(a)、HA@SA(b)和HA@SA-Cr(Ⅵ)(c)的FT-IR 光谱
2.1.2 Zeta电位
由图2 可知,随着pH 的增大,微球悬浮液的Zeta电位都逐渐下降。在pH 约为2.12 时,SA 微球处于零点电荷(ZPC);在pH 约为5.04 时,HA@SA 微球处于ZPC。表明在pH 小于5.04 时,HA@SA 微球表面带正电荷,这对吸附以负离子形式存在的六价铬离子有利;在pH 大于5.04时,HA@SA 微球表面带负电荷,有利于解吸六价铬离子。这与Lin等[9]的研究结果一致。

图2 不同pH 下SA、HA@SA 微球的Zeta 电位
2.1.3 EDS
由图3a 可知,SA 微球表面存在C、O、Ca 元素。由图3b 可知,HA@SA 微球表面除了C、O、Ca 外,还检测到N,表明HA 已成功接枝到SA 表面。由图3c 可知,吸附Cr(Ⅵ)后,HA@SA 微球表面除了C、O、N、Ca 外,还检测到Cr,表明Cr(Ⅵ)已被成功吸附到HA@SA 微球表面。HA@SA 微球对Cr(Ⅵ)的吸附主要源于HA 中质子化的胺基和阴离子Cr(Ⅵ)(存在形式HCrO4-和Cr2O72-)之间的静电吸引。

图3 SA(a)、HA@SA(b)和HA@SA-Cr(Ⅵ)(c)的能量色散谱图
2.1.4 XPS
由图4a 可知,SA 微球表面存在C、O 元素峰;由图4b 可知,HA@SA 微球表面除了C、O 元素峰外,还出现了N 元素峰(来自HA 中的胺基和亚胺基官能团),表明HA 被成功接枝到SA 表面,这与FT-IR 和EDS 结果一致;由图4c 可知,吸附Cr(Ⅵ)后,HA@SA微球表面除了C、O 和N 元素峰外,还出现了Cr 2p峰,表明Cr(Ⅵ)被HA@SA 微球成功吸附。

图4 SA(a)、HA@SA(b)和HA@SA-Cr(Ⅵ)(c)的XPS 总能量光谱图
2.2 影响HA@SA 微球吸附Cr(Ⅵ)的因素
2.2.1 pH
pH 影响重金属离子在溶液中的存在形式、溶解状况以及吸附剂表面的电荷分布情况,进而决定吸附剂与重金属离子的相互作用[11]。由图5 可知,随着pH 的增大,HA@SA 微球对Cr(Ⅵ)的吸附量先增加后降低;当pH 为2 时,吸附量达到最大。pH 从1 增大到2 时,Cr(Ⅵ)的存在形式从H2CrO4向HCrO4-和Cr2O72-转化[8]。在酸性条件下,HA 中的胺基大量质子化,Cr(Ⅵ)以阴离子(HCrO4-和Cr2O72-)形式通过静电作用吸附于带正电荷的吸附剂上,实现溶液中Cr(Ⅵ)的去除。因此,为了去除Cr(Ⅵ)并最大程度地发挥吸附剂的作用,后续吸附实验溶液pH 选择2。另外,随着pH的增大,胺基从质子化状态转变为去质子化;进一步提高pH,吸附剂表面带负电荷,与HCrO4-和Cr2O72-之间存在静电斥力,使HA@SA 微球对Cr(Ⅵ)的吸附量大大降低。因此,解吸过程在碱性条件下进行有利。

图5 pH 对HA@SA 微球吸附Cr(Ⅵ)的影响
2.2.2 Cr(Ⅵ)初始质量浓度与吸附等温线
在间歇吸附过程中,重金属离子初始质量浓度作为克服溶液与固相吸附剂之间传质阻力的推动力起着关键作用[12]。由图6 可知,HA@SA 微球的吸附量大大高于SA 微球,这是因为HA@SA 微球上的吸附位点更多。随着Cr(Ⅵ)初始质量浓度的增加,HA@SA微球的吸附量也随之增大,随后吸附达到饱和。这是因为当Cr(Ⅵ)初始质量浓度较小时,吸附剂表面的吸附位点足以吸附溶液中的Cr(Ⅵ)。随着Cr(Ⅵ)初始质量浓度的增加,HA@SA 微球对Cr(Ⅵ)的吸附量呈明显上升的趋势。这是因为增大Cr(Ⅵ)初始质量浓度,提高了HCrO4-和Cr2O72-与微球表面之间的质量浓度梯度,增大了吸附的推动力,促使更多的HCrO4-和Cr2O72-扩散、吸附到HA@SA 微球表面。随着Cr(Ⅵ)初始质量浓度的进一步增大,HA@SA 微球上的位点吸附饱和,吸附值基本保持不变。

图6 Cr(Ⅵ)初始质量浓度对SA、HA@SA 微球吸附Cr(Ⅵ)的影响
为了进一步探索HA@SA 微球对Cr(Ⅵ)的最大吸附量并描述该吸附过程,用等温线分析HA@SA 微球吸附Cr(Ⅵ)的平衡数据。本研究通过模拟Langmuir、Freundlich 两种平衡模型来研究吸附等温线。
Langmuir 模型基于吸附剂表面结构均匀的假设,其中所有的吸附位点相同,并且在能量上相等[9]:
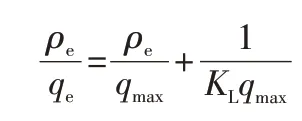
其中,qe是平衡时的金属离子吸附量,mg/g;ρe是平衡时的金属离子质量浓度,mg/L;qmax是理论最大吸附量,mg/g;KL与吸附能有关,L/mg。
Freundlich 模型是描述非均相体系的经验方程。假定吸附质分子在吸附剂表面形成多层吸附,相邻吸附质分子之间具有相互作用[9]:
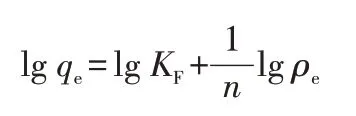
其中,KF和n是Freundlich 常数;ρe是平衡时的金属离子质量浓度,mg/L。
由表1中的相关系数R2可看出,Freundlich模型比Langmuir 模型具有更好的拟合效果,表明Freundlich模型能更好地描述Cr(Ⅵ)被吸附到HA@SA 微球表面的过程,即Cr(Ⅵ)可能以多层形式被吸附到HA@SA微球表面。由Langmuir 模型计算SA 微球对Cr(Ⅵ)的理论最大吸附量仅为24.86 mg/g,而HA@SA 微球为247.50 mg/g,与SA 微球相比提高了8.96 倍,这得益于HA 中含有大量的胺基官能团。

表1 SA、HA@SA 微球吸附Cr(Ⅵ)的等温线参数
2.2.3 吸附时间与吸附动力学
在吸附剂的使用过程中,吸附时间既反映了吸附过程的经济性,又反映了吸附过程的效率[12]。吸附时间对HA@SA微球吸附Cr(Ⅵ)的影响见图7。

图7 吸附时间对HA@SA 微球吸附Cr(Ⅵ)的影响
由图7 可知,Cr(Ⅵ)的吸附量随着吸附时间的延长而增加,26 h后基本达到平衡。在吸附初始阶段,吸附速率相对较快,主要是因为HA@SA 微球上存在大量空的吸附位点,而且在溶液和吸附剂表面之间存在较大的Cr(Ⅵ)质量浓度梯度,吸附推动力很大。随着时间的延长,HA@SA 微球上吸附位点不断被占据,Cr(Ⅵ)质量浓度梯度降低,导致吸附速率减小。因此,后续吸附实验吸附时间选择26 h。
吸附动力学对于HA@SA 微球去除Cr(Ⅵ)的实际应用至关重要,有助于理解吸附机理及其可能的控制步骤[13]。参考相关文献,通过4 种模型来研究吸附过程的动力学。
准一级动力学模型是描述固体吸附剂在水溶液中吸附过程最常用的模型之一[13]:
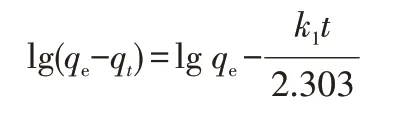
其中,k1是准一级动力学模型的速率常数,h-1;qe和qt分别是平衡和时间为t时的吸附量,mg/g。
准二级动力学模型基于吸附速率受化学吸附机理控制的假设,这种化学吸附涉及吸附剂与吸附质之间的电子共用与电子转移[13]:

其中,k2是准二级动力学模型的速率常数,g/(mg·h)。
Elovich 动力学模型描述固体在水介质中对吸附质的吸附[13]:

其中,α是Elovich 方程的初始吸附速率,mg/(g·h);β与化学吸附的表面覆盖度和活化程度有关。
韦伯-莫里斯动力学模型又被称为颗粒内扩散模型,可以更好地识别涉及到的扩散机制[12]:

其中,kpi是阶段i的颗粒内扩散速率常数,mg·h0.5/g;dpi是阶段i的截距,截距越大,边界层效应越大。
动力学模型拟合参数计算结果见表2。

表2 HA@SA 微球吸附Cr(Ⅵ)的动力学参数
比较准一级、准二级、Elovich 动力学模型的拟合结果,准二级动力学吸附模型的R2优于准一级、Elov⁃ich 动力学吸附模型;此外,准二级动力学吸附模型的平衡吸附量接近实验值,表明吸附过程符合准二级动力学吸附模型,吸附过程主要由化学吸附控制。对于颗粒内扩散模型,当拟合直线通过原点时,吸附过程仅由颗粒内扩散控制;否则吸附过程涉及多个扩散阻力[6]。
由表2 可知,截距dpi不为零,这意味着颗粒内扩散不是吸附过程唯一的速率控制步骤。
2.2.4 温度与吸附热力学
温度是吸附剂吸附重金属离子的重要参数,影响固-液界面、吸附剂的溶胀性和重金属离子的流动性[12]。由图8 可以看出,吸附量随着温度升高而增大,表明HA@SA 微球吸附Cr(Ⅵ)是吸热过程。而且吸附量随温度升高增加不显著。从节能角度考虑,后续实验的吸附温度选择30 ℃。

图8 温度对HA@SA 微球吸附Cr(Ⅵ)的影响
吸附过程的热力学参数反映吸附过程的性质,即自发性、随机性、吸热性或放热性[12]。用下列公式计算HA@SA 微球吸附Cr(Ⅵ)的热力学参数:
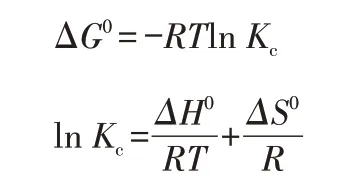
其中,R是摩尔气体常数,8.314 kJ/(mol·K);T是热力学温度,K。Kc可定义为:

其中,ρs为吸附到吸附剂上的Cr(Ⅵ)质量浓度,mg/L;ρ0是Cr(Ⅵ)初始质量浓度,mg/L;ρe是平衡时溶液中的Cr(Ⅵ)质量浓度,mg/L。
由表3 可知,吉布斯自由能变ΔG0均为负值,证实了吸附过程的可行性和自发性。根据Van′t Hoff 方程的斜率和截距计算了焓变ΔH0和熵变ΔS0,由表3可知,ΔH0为正值,表明HA@SA 微球吸附Cr(Ⅵ)的过程吸热。ΔS0为正值,表明在吸附过程中水合球释放水分子,使得Cr(Ⅵ)结合到固-液界面的随机性增加。

表3 HA@SA 微球吸附Cr(Ⅵ)的热力学参数
2.3 再生性能
可重复使用性是良好吸附剂的重要特征[14]。由图9 可知,经过8 次吸附-解吸循环实验后,HA@SA微球对Cr(Ⅵ)的吸附能力仍保持在80%以上,表明HA@SA 微球具有良好的再生性能。吸附能力降低可能是由于HA@SA 微球在吸附-解吸过程中质量损失或解吸不完全。
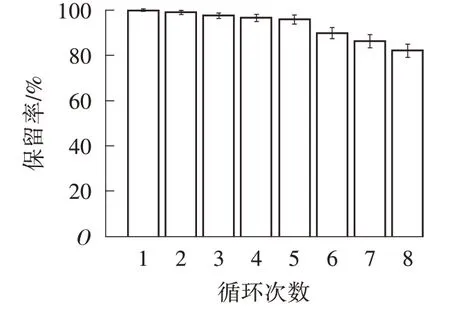
图9 HA@SA 微球对Cr(Ⅵ)吸附的可重复使用性
3 结论
(1)以自制的超支化聚酰胺为改性剂,戊二醛为交联剂,将HA 接枝到SA 微球表面,成功制备了超支化聚酰胺改性海藻酸钠微球吸附剂。
(2)HA@SA 微球吸附Cr(Ⅵ)的最佳pH 为2,吸附过程符合Freundlich 等温线模型,Cr(Ⅵ)以多层形式被吸附到非均相HA@SA 微球表面。HA@SA 微球理论最大吸附量达247.50 mg/g,比SA 微球(24.86 mg/g)提高了8.96 倍。HA@SA 微球吸附Cr(Ⅵ)与准二级动力学模型有较好的拟合效果,表明吸附过程主要被化学吸附控制,而吸附过程的速率控制步骤也不只是颗粒内扩散,吸附过程是自发的吸热过程。
(3)经过8 次连续的吸附-解吸循环实验,吸附剂的吸附能力能保持在80%以上。
