短肋多指综合征3型一家系的临床表型和DYNC2H1基因突变分析
2020-09-16张建芳陈必良
詹 瑛,张建芳,程 璐,徐 盈,陈必良
(1解放军第63750部队医院妇产科,西安 710043;2空军军医大学第一附属医院妇产科;*通讯作者,E-mail:zhzhhao@163.com)
骨骼发育不良是一类异质性很大的遗传性疾病,主要与软骨和骨骼发育过程异常有关。国外的一项研究报道指出目前有226个基因与456种已知的遗传性骨骼发育不良相关[1]。短肋多指综合征(short-rib polydactyly syndromes, SRPSs)就是其中的一组骨骼发育不良症候群,是由纤毛功能紊乱导致的以肋骨短小、长骨短小、多指(趾)畸形,伴多脏器损害为特征的一种罕见的常染色体隐性单基因遗传病。根据国际骨软骨发育不良分类,已识别出4种不同类型的SRPS[2]。其中短肋多指综合征3型(short-rib polydactyly syndrome type Ⅲ,SRPS Ⅲ)是其中表型最重、预后较差的一种,往往妊娠中期以后宫内即有影像学的异常表现。尽管影像学技术现今取得了巨大的进步,但胎儿骨骼发育不良数量众多且表型特征往往多有重叠,故而仍然难以实现精准的产前诊断。鉴于该类疾病各种形式的广泛的遗传型和表现型异质性,基于传统Sanger测序确定可能的致病基因是不切实际、费时费力的。因此,基于二代测序(NGS)技术的靶向捕获、全外显子测序、全基因组测序等为识别遗传性骨骼疾病的致病突变提供了一种新的高效的方法。
本研究基于NGS采用全外显子测序(whole exome sequencing,WES)的方法对一个SRPS Ⅲ家系引产胎儿进行全外显子测序,数据分析结果显示该先证者的DYNC2H1基因存在复合杂合突变c.2346-1G>A和c.7292+4T>C,再结合PCR和Sanger测序在该家系中进行验证,先证者两处变异分别遗传自其父母,符合SRPS Ⅲ常染色体隐性遗传的规律。从遗传学的角度初步明确了该家系中先证者SRPS Ⅲ的可能发病原因,为其临床诊断提供了有力的证据,为该家系的遗传咨询和产前诊断提供了可靠的分子依据。
1 资料与方法
1.1 临床资料
该家系中母亲孕2产0,2016年及2017年均因“胎儿四肢极度短小”于孕中期引产。夫妻双方非近亲结婚,否认双方家族中遗传病史。2016年6月第1次妊娠,孕早期经过顺利,孕13周时西安市第四医院产科超声提示胎儿双肺回声增强、胎儿四肢形态异常,未见明确长骨回声,腹腔囊性包块,鼻骨显示不清;连续4周超声动态监测胎儿骨骼系统发育情况仍提示胎儿四肢形态异常,成骨发育不全可能。该家系决定终止妊娠,由于当时就诊医院客观条件受限,引产胎儿未做尸检,未留取样本进一步行基因检测。2017年4月该家系中母亲第2次妊娠,孕12周西安市第四医院产科超声检查提示胎儿全身皮肤水肿,头颈部淋巴水囊瘤,建议加强产检、进一步行产前诊断。孕17周时复查超声提示胎儿颈部水囊瘤,双肾盂重度积水,胎儿四肢长骨短小、双手可见多指,胎儿心脏异常,建议进一步胎儿心动超声及产前诊断。该孕妇及家属坚决要求终止妊娠,且为求进一步遗传咨询转往西京医院。于孕17+4周时引产,引产胎儿为男性体征,查体可见全身皮肤轻度水肿,四肢明显短小,双手六指畸形,余外观未见明显异常。留取引产胎儿脐带组织行基因检测。
1.2 基因组DNA提取
该家系夫妻双方知情同意,签署知情同意书后,采用EDTA抗凝管抽取该夫妻双方静脉血各5 ml,按血液基因组DNA提取试剂盒(北京天根生物科技有限公司)提取外周血基因组DNA,-20 ℃保存备用。
1.3 全外显子测序
将该家系中二胎患病胎儿的新鲜脐带组织5 g左右,剔除结缔组织,吸水纸吸干血液,剪碎后放入研钵,倒入液氮磨成粉末,按DNA提取试剂盒(北京天根生物科技有限公司)说明书提取胎儿DNA,送至北京金准医学检验所,利用Agilent SurSelect人全外显子组芯片捕获试剂盒构建文库,使用Illumina Hiseq X Ten测序平台进行二代测序,数据经过质量控制后与基因组参考序列进行比对,查阅相关数据库综合分析判断可疑变异的致病性。
1.4 PCR和Sanger测序验证
根据全外显子高通量测序的结果,用PCR结合Sanger测序对二胎胎儿及其父母进行突变检测和验证。针对DYNC2H1基因的c.2346-1G>A和c.7292+4T>C位点采用Primer5.0软件设计跨越16号、44号外显子上下游的引物序列。DYNC2H1-16F:TGGCTTAGCAACTGTAGAAGCA,16R:AGCCCGGATTTCTTCAAAAG;DYNC2H1-44F:TTGACGTATCAAGGATTTTATG,44R:CTGTTCATACACTTGTACCAT。引物由上海生工生物工程有限公司合成。PCR反应条件为:95 ℃ 10 min,95 ℃ 30 s,60 ℃ 30 s,72 ℃ 45 s,共35个循环,最后72 ℃延伸5 min。PCR产物用1%琼脂糖凝胶电泳检测,之后进行双向测序。
2 结果
2.1 全外显子高通量测序结果
引产胎儿基因组DNA高通量测序数据分析发现胎儿DYNC2H1基因存在c.2346-1G>A变异和c.7292+4T>C变异两处杂合突变(见图1),两处变异在人群dbSNP数据库、HGMD数据库、ClinVar数据库中均未见报道。查阅HGMD与ClinVar数据库,DYNC2H1基因据报道与短肋多指综合征相关。短肋多指综合征为常染色体隐性遗传病,上述两处变异均为DYNC2H1基因内含子区域的变异,其中c.2346-1G>A位点变异位于16号内含子-1位,紧邻17号外显子,处于“经典剪接区域”,该区域的碱基突变一般均会对原剪切位点产生影响;c.7292+4T>C位点变异位于44号内含子+4位,紧邻44号外显子,紧挨“经典剪接区域”,该区域的碱基突变一般均会对原剪切位点产生影响。DYNC2H1基因上述两个位点的变异高度怀疑会影响转录水平内含子序列的剪切,进而影响DYNC2H1基因所编码的蛋白质功能。利用Human Splicing Finder软件(http://www.umd.be/HSF/)对上述两个内含子区域位点进行生物信息学的在线预测,结果提示,c.2346-1G>A位点对于剪切影响的可能超过70%,c.7292+4T>C位点可能不影响原有的剪接方式(见图2)。

图2 HSF生物信息学软件预测DYNC2H1基因变异位点的影响Figure 2 The predicted influence of DYNC2H1 gene mutations through HSF software

图1 胎儿DYNC2H1基因变异位点在基因序列中的位置Figure 1 The locations of the fetal mutations on DYNC2H1 gene sequencing
2.2 Sanger测序验证结果
Sanger测序结果显示,患病胎儿DYNC2H1基因c.2346-1G>A、c.7292+4T>C的复合杂合突变分别来自该家系中的母亲及父亲,其父母分别为上述杂合突变的携带者,该遗传方式符合常染色体隐性遗传(见图3),如果上述两处变异为致病性突变,该家系再次妊娠有1/4的概率生育患者。
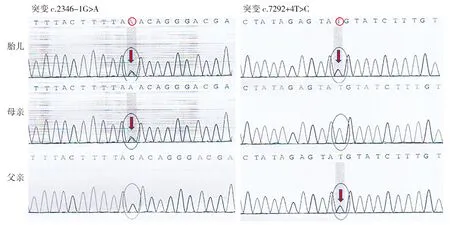
图3 该家系中DYNC2H1基因变异Sanger测序验证结果Figure 3 The validation of the mutations of DYNC2H1 gene in the affected family
3 讨论
遗传性骨病是指由于遗传物质改变导致骨骼畸形的一类遗传性疾病,它是一大类具有遗传异质性和表型异质性的骨软骨发育不良病。这类疾病大多是发病率很低的罕见病,但种类繁多,总发病率大于1/5 000[3]。这些骨病发病早,症状明显,以骨骼塑形、生长、分化、内稳态异常为主要特征,通常造成患者致残甚至致死,其发病率与死亡率相当可观,而且此类骨病还会逐代或隔代遗传,故危害十分严重,往往给家庭和社会带来沉重的经济负担。短肋多指综合征(SRPSs)是其中的一种常染色体隐性遗传性骨病。虽然SRPSs影像学表现往往以肋骨短小、长骨短小、多指(趾)畸形、伴多脏器损害为特征,但其病情轻重程度、病因及预后往往多种多样,而且常在胎儿发育期间宫内即有表现,产前诊断SRPSs难度很大,许多时候孕妇在尚未查明病因时便选择了终止妊娠,无法为再次妊娠提供产前诊断的可靠依据。根据国际骨软骨发育不良分类,四种不同类型的SRPSs已被确认,分别为SRPS Ⅰ型(Saldino-Noonan综合征)、SRPS Ⅱ型(Majewski综合征)、SRPS Ⅲ型(Verma-Naumoff综合征)、SRPS Ⅳ型(Beemer-Langer综合征),其中SRPS Ⅰ型和SRPS Ⅲ型目前已被认为是同一类型,也是表现最为严重的一种类型[2,4]。目前与SRPS Ⅲ型相关的致病基因均属于鞭毛细胞内运输(IFT)相关基因,如DYNC2H1、IFT80、WDR34、WDR35、WDR60等[5-8]。
本研究中孕妇连续两胎均出现相同的临床表型,遗传性骨骼疾病和单基因病的可能性非常大[9]。本课题组用家系全外显子组测序检测发现胎儿DYNC2H1基因存在复合杂合突变(c.2346-1G>A变异和c.7292+4T>C变异)。DYNC2H1基因Sanger测序验证结果显示,胎儿c.2346-1G>A和c.7292+4T>C分别来自健康的母亲和父亲,符合常染色体隐性遗传方式。DYNC2H1基因位于染色体11q22.3,长13 kb,有90个外显子。目前我国仅有数例散发SRPS Ⅲ型病例报道是由DYNC2H1基因突变所致[10-13],而且具有明显的遗传异质性和表型多样性。DYNC2H1基因编码胞质动力蛋白亚基重链,是纤毛内转运蛋白复合体A的组成部分,在逆向转运过程中,纤毛内转运蛋白复合体A与胞内信号分子结合,胞质动力蛋白亚基与纤毛顶部的双微管结合,并沿着双微管从纤毛顶部向底部移动,并参与纤毛蛋白的循环利用。表明DYNC2H1蛋白在哺乳动物纤毛的延伸组成和形态维持方面都具有关键的作用,而纤毛是软骨内骨发育过程中的重要组成部分[14]。纤毛结构广泛分布于真核动物的各类细胞中,其结构进化高度保守,所以DYNC2H1基因的致病性变异表现为以软骨、骨骼成骨异常为主要表型特征,伴随多系统畸形的一组异型性疾病。Schmidts等[15]检测到DYNC2H1基因敲除影响秀丽隐杆线虫和小鼠的纤毛内转运蛋白复合体逆向转运功能,导致纤毛发育异常,并观察到与骨骼发育紧密相关的Hedgehog信号通路出现异常调控,导致骨骼发育缺陷。SRPS Ⅲ型是一种典型的骨骼纤毛疾病,初级纤毛和胞质动力蛋白是骨骼发育过程中形态发生途径的必要组成部分,细胞纤毛运输蛋白在软骨细胞的成熟方面具有非常重要的作用[16]。
截止2020年4月,ClinVar数据库已报道DYNC2H1基因致病或可能致病的变异位点有236个(https://www.ncbi.nlm.nih.gov/clinvar/),而本研究发现的两处可疑致病位点尚无文献报道。研究表明DYNC2H1基因庞大尚没有热点突变,这些突变基本都是复合杂合突变,但到目前为止并没有发现纯合突变,没有纯合子的形成可能是因为该基因过于保守和关键,纯合子的早期胚胎已死亡[12]。本研究中发现的c.2346-1G>A变异和c.7292+4T>C变异均位于DYNC2H1基因内含子区域,生物信息学软件借助特定算法预测其一高度可能影响剪切受体,其一可能不影响原有剪切,但是否真正影响剪切只有通过分子生物学实验进一步验证才能确定。综上所述,对于该家系课题组利用WES技术在该家系中筛选出了两个高度可疑的DYNC2H1基因致病位点,且在家系中存在遗传共分离现象,结合临床表型分析,上述两个位点很可能是该家系的致病位点,建议家系再次妊娠之前完成其致病性的验证,可以定制Minigene验证上述两个位点对DYNC2H1基因转录水平的影响,进而分析其致病性。若为致病性变异,则可以根据上述两个位点进行产前诊断或是胚胎植入前遗传学诊断,从而实现SRPS Ⅲ型患者在家族中的阻断,指导家系成员实现优生优育。
近些年来二代测序技术已经广泛应用于临床疾病的基因突变检测,研究者可以根据基因突变的特点与其他传统的检测技术结合使用,相互配合取长补短,帮助临床上更加经济、精准、快速地诊断疾病。遗传性骨病的致病基因种类繁多,每个基因的致病位点动辄上百,如果用Sanger测序逐一排查经济效率极其低下,所以本研究选择NGS和Sanger测序相结合的方法进行基因诊断,可以快速准确找到致病基因及致病位点,大大提高检测效率。
