Diffuse alveolar hemorrhage with histopathologic manifestations of pulmonary capillaritis:Three case reports
2020-09-15
Jun Xie,Ying-Yue Zhao,Jing Liu,Guang-Min Nong,Department of Pediatrics,the First Affiliated Hospital of Guangxi Medical University,Nanning 530021,Guangxi Zhuang Autonomous Region,China
Abstract
Key words:Diffuse alveolar hemorrhage;Pulmonary capillaritis;Glucocorticoid;Immunosuppressant;Lung biopsy;Case report
INTRODUCTION
Diffuse alveolar hemorrhage (DAH) is a rare,and life-threatening disease in children.It is reported that DAH in children is associated with common causes such as oligoimmunity,immune complex deposition,and others (mainly due to medication)[1].DAH is divided into DAH in the presence or absence of pulmonary capillaritis (PC),and DAH of cardiac origin[2].PC is characterized by the presence of cough,shortness of breath,dyspnea,with or without hemoptysis[3],and iron deficiency anemia.Histopathology of PC is manifested as neutrophil aggregation or infiltration at the tight junction of alveolar interstitial and alveolar capillaries[4],as well as nuclear debris.Red blood cells and hemosiderin-laden macrophage cells (HLMs)exuded from capillaries are seen in the alveolar spaces and interstitium.In this report,we describe three patients with PC diagnosed by lung biopsy.
CASE PRESENTATION
Case 1
A girl aged 6 years and 3 mo presented with repeated pale skin and cough for>2 years.She was allergic to chicken protein and milk and denied medical history and family genetic diseases.Hemoglobin was 109 g/L,erythrocyte sedimentation rate(ESR) was 48 mm/h,and urinalysis,renal function,coagulation function and serum ferritin were normal.Antinuclear antibody (ANA) (IgG) was positive,immunofluorescence revealed granular type (1:320),and the other tests were negative.Chest high-resolution computed tomography (HRCT) showed patchy density and ground-glass opacity in multiple lobes (Figure1A).A right upper lung biopsy showed typical DAH and PC (Figure2A).
Case 2
A girl aged 11 years and 6 mo was admitted to hospital with a 3-year history of recurrent of cough and hemoptysis.She previously had episodes of hemoptysis for which she had received multiple treatments,including antibiotics and glucocorticoids.Laboratory examination revealed hemoglobin 75 g/L.Evaluation was normal for ESR,urinalysis,renal function,ANA,and anti-glomerular basement membrane antibody(GBM).Chest HRCT showed diffuse cystic lesions in bilateral lung fields (Figure1B).There were no malignant cells in the bronchoalveolar lavage smear,and HLMs were visible.A diagnosis of PC was confirmed on right lung biopsy that showed spilled red blood cells and HLM aggregation,and a small amount of lymphocyte and neutrophil infiltration,typical of DAH and PC (Figure2B).
Case 3
A girl aged 11 years and 11 mo presented to our hospital with a history of recurrent pale skin and hemoptysis for>1 year.The child had no significant past medical problems,and had no exposure to toxic chemicals,fumes,or other irritants.Auxiliary examination showed hemoglobin 79 g/L.She was admitted;evaluation included normal results for platelets;and ESR,urinalysis,renal function,coagulation function,ANA,and antineutrophil cytoplasmic antibodies were within the normal range.Chest HRCT showed diffuse ground-glass opacity in the bilateral lung fields.Histopathology of the lung biopsy was typical of DAH,but the lesions were mainly in the chronic phase (fibrosis,lymphatic,and plasma cell infiltration) (Figure2C).
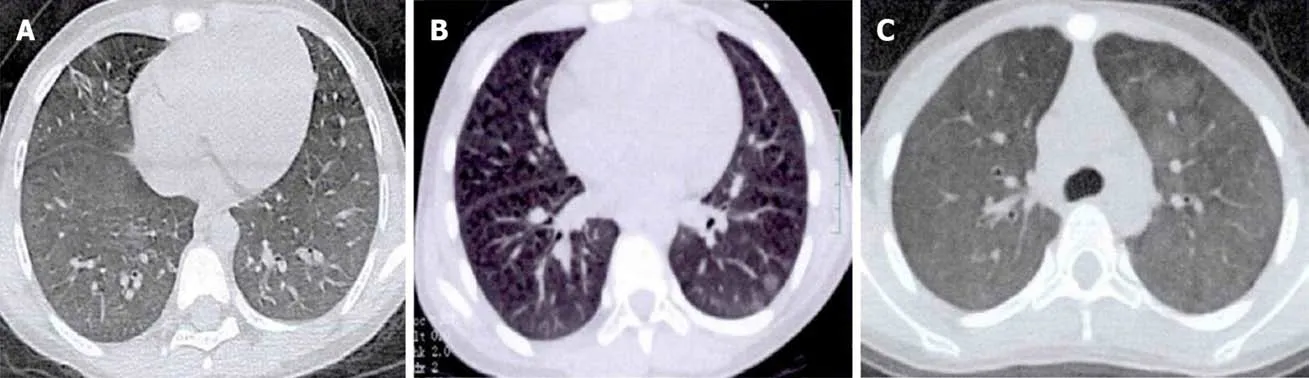
Figure1 Chest high-resolution computed tomography.A:Reduced lucency of both lungs,patchy density and ground-glass opacity were seen in multiple lobes with fuzzy boundaries;B:Diffuse cystic lesions in both lungs and patchy opacity with fuzzy boundaries in multiple lobes;C:Diffuse ground-glass opacity was observed in both lungs with fuzzy boundaries.
FINAL DIAGNOSIS
Case 1 was positive for ANA (IgG) and immunofluorescence test revealed granular IgG,and a small amount of granular IgA deposit on the alveolar wall combined with minimal change glomerulonephritis,which suggested systemic lupus erythematosus(SLE).Cases 2 and 3 were diagnosed with isolated pauci-immune PC (IPPC),which was based on histopathological evidence of PC in the absence of clinical and serological evidence of an underlying systemic disorder.
TREATMENT
Case 1
The patient was given oral prednisone 10 mg/d (1.58 mg/kg/d) with subsequent gradual taper plan over 3 mo.
Case 2
The child was treated with intravenousmethylprednisolone 54 mg q12 h (4 mg/kg/d)for 5 d,follow by oral prednisone at a dose of up to 30 mg/d (1 mg/kg/d),and this relieved her symptoms of cough,hemoptysis,and shortness of breath.
Case 3
The patient was treated with prednisone 30 mg/d (1.2 mg/kg/d) for 4 mo,gradually reduced to 15 mg/d (5 mg every 2 mo) for 9 mo.This resulted in symptom improvement of the pulmonary hemorrhage.
OUTCOME AND FOLLOW-UP
Case 1
The patient attended regular outpatient review,and symptoms were improved.Hemoglobin was 128-147.8 g/L;ESR WAS 29 mm/h;chest HRCT showed that the lung lesions improved;and percutaneous kidney biopsy was performed and revealed minimal change glomerulonephritis (Figure3).Oral prednisone was gradually reduced (5 mg every 2-3 mo),and 14 mo later,the prednisolone dose was tapered from 5 mg/d to a maintenance dose of 5 mg every other day.Follow-up has been completed for 2 years with no evidence of recurrence after discontinuation of prednisone.
Case 2
Follow-up hemoglobin concentration fluctuated at 117.4-128.8 g/L;ESR was normal;chest HRCT showed partial absorption of inflammatory lesions,and the remaining lesions were similar to before.After oral administration of prednisone for 2 years,the dose was reduced to 5 mg/d.During that period,the patient was hospitalized twice for DAH,and the dose of prednisone was increased to 60 mg/d (1.5 mg/kg/d),and methotrexate was added at 12.5 mg once weekly.Four years after the initial diagnosis,the child remained on therapy and the symptoms had resolved,but she developed Cushing’s syndrome.
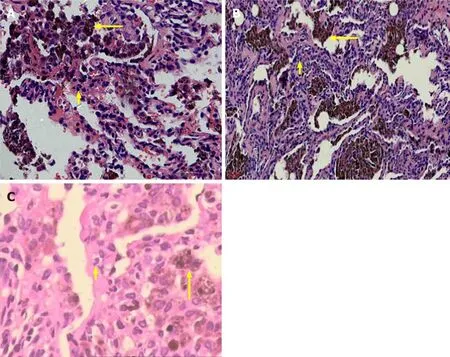
Figure2 Hematoxylin and eosin staining.A:Aggregation of hemosiderin-laden macrophage cells (HLMs) within the alveolar compartment (long arrow),alveolar interval are mild fibrous thickening and neutrophil infiltrate (short arrow) (400 ×);B:Acute extravasation of fibrin and red blood cells,HLM deposits in the alveolar cavities (long arrow) and neutrophil infiltrates (short arrow) (400 ×);C:Partial lung tissue fibrosis and HLM deposits were observed in the alveolar cavities (long arrow)and neutrophil infiltrates (short arrow) (400 ×).
Case 3
The patient had regular follow-up visits,and she had no cough or hemoptysis,no pale skin.Routine blood examination showed hemoglobin 112.6-140 g/L,and ESR,routine urinalysis and anti-GBM antibody were negative.Chest HRCT showed that the lung lesions were not aggravated compared with before.The patient has been stable and asymptomatic for 3 years.
DISCUSSION
PC is a histopathological type of DAH that is characterized by inflammatory cell infiltration and blood accumulation in the alveolar cavity[5,6].Two of the present cases were diagnosed with IPPC,which was based on histopathological evidence of PC in the absence of clinical and serological evidence of an underlying systemic disorder[3].In Case 1,ANA autoantibodies (IgG) were positive and immunofluorescence test revealed granular IgG,a small amount of granular IgA deposit on the alveolar wall,combined with minimal change glomerulonephritis,which suggested SLE[7,8].Only 5%of SLE patients reportedly have DAH,but PC,IgG,and complement C3 immune complex deposits are observed microscopically in nearly 50% of cases[8].There is no unified diagnostic standard for SLE with PC.The clinical symptoms of most PC patients are inconspicuous,and lung pathology is the gold standard for diagnosis.
Currently,glucocorticoids alone or combined with immunosuppressants to control inflammation and autoimmune response remain the standard treatment for most patients.It is reported that glucocorticoids (starting with intravenous methylprednisolone 2-4 mg/kg/d for 1-3 d,followed by oral prednisone 1-2 mg/kg/d;or starting with oral prednisone 1-2 mg/kg/d)[1]are effective,but fail in some cases.When glucocorticoids are ineffective,immunosuppressants are recommended,including oral methotrexate,hydroxychloroquine,and azathioprine[9-11].After addition of immunosuppressants,the condition in most cases can be controlled[9,10].Rituximab and recombinant-activated human factor VIIa seem to be a new promising therapy[3,12].
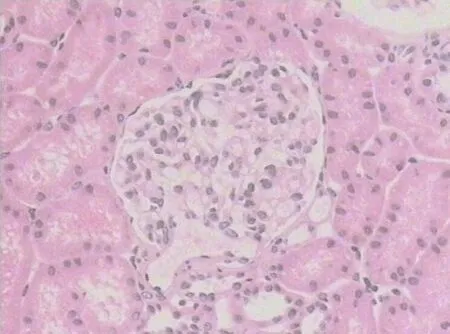
Figure3 Hematoxylin and eosin staining.Twenty nonsclerotic glomeruli were identified by light microscopy,which mainly presented as mesenteric proliferative changes.No glomeruli contained fibrous crescents and no proliferative changes were observed in the endocapillary or extracapillary areas (400 ×).
CONCLUSION
PC is classified as isolated PC and immune-mediated PC associated with systemic disease.The condition can be controlled in most children using glucocorticoids alone or in combination with immunosuppressants.
杂志排行

World Journal of Clinical Cases的其它文章
- Assessment of diaphragmatic function by ultrasonography:Current approach and perspectives
- Computer navigation-assisted minimally invasive percutaneous screw placement for pelvic fractures
- Research on diagnosis-related group grouping of inpatient medical expenditure in colorectal cancer patients based on a decision tree model
- Evaluation of internal and shell stiffness in the differential diagnosis of breast non-mass lesions by shear wave elastography
- Real-time three-dimensional echocardiography predicts cardiotoxicity induced by postoperative chemotherapy in breast cancer patients
- Lenvatinib for large hepatocellular carcinomas with portal trunk invasion:Two case reports