高分辨液质联用法测定奥司他韦原料药中有关物质*
2020-09-10陈志谦符旭东钟昌平
陈志谦,符旭东,钟昌平
(1. 湖北省武汉市汉阳医院,湖北 武汉 430050; 2. 中国人民解放军中部战区总医院,湖北 武汉 430070)
奥司他韦磷酸盐是一种用于治疗流感的抗病毒药物,可选择性抑制病毒的神经氨酸酶来控制病毒的复制,用于治疗流感。与扎那米韦(zanamivir)相比,奥司他韦属乙酸酯类药物,在体内肝酯酶作用下能迅速转化为高活性的奥司他韦羧酸盐,有利于生物体的快速吸收,可大大减少并发症的发生(如气管与支气管炎、肺炎等)[1-2],是目前治疗禽流感最有效的药物[3-4]。随着禽流感在全球范围的暴发,奥司他韦作为对H1N1,H5N1,H9N2 等亚病毒具有抑制作用的抗病毒药物受到广泛关注。由于流行病暴发及相应市场需求的无法预测性,大量的奥司他韦会被存储起来,以防备流行病的发生,超过5 年保存期导致大量过期,又因其存储昂贵而未被销毁。关于过期奥司他韦的药物有效性及安全性,欧洲药品管理局和美国食品药物管理局(FDA)规定,在非特殊情况下,不得使用过期及未按要求储存的奥司他韦。同时,学术界对过期奥司他韦的安全性及成分也开展了研究[5-6]。本研究中采用超高效液相色谱-四极杆-飞行时间串联质谱(UPLC-Q-TOF-MS)法测定奥司他韦原料药中的有关物质,并对其结构进行鉴定,旨在为奥司他韦的质量控制及安全使用提供依据。现报道如下。
1 仪器与试药
1.1 仪器
Waters acquity 液相色谱仪,Xevo TQ-S 型质谱仪,配备电喷雾离子源,飞行时间质谱检测器,Mass Lynx V4.1 数据分析软件,均购自美国沃特世公司;Mettler AE-240 型电子天平(梅特勒公司,精度为0.1 mg)。
1.2 试药
奥司他韦对照品(中国食品药品检定研究院,批号为101096-201501);奥司他韦原料药(药企A,批号为180621-34);甲酸为LCMS 级,乙腈为色谱纯,水为超纯水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱与质谱条件
液相色谱条件:色谱柱为Kinetex 柱(100mm×2.1mm,2.6 μm);流动相为0.1%甲酸水溶液(A)-乙腈(B),梯 度 洗 脱(0~1 min 时2%B,7~9 min 时100%B,9.1~12 min 时2%B);流速为0.4 mL / min;检测波长为207 nm;柱温为40 ℃;进样量为5 μL。
质谱条件:ESI 离子源,正离子全扫描方式,MS1 扫描范围为质荷比(m/ z)100~1 000,MS2 扫描范围为m / z 100~1 000;喷雾电压为2.5 kV;锥孔电压为25 V;去溶剂温度为500 ℃;去溶剂气体为800 L/h。
2.2 溶液制备
取奥司他韦对照品5 mg,精密称定,置5 mL 容量瓶中,加甲醇溶解并定容,摇匀,滤过,即得质量浓度为1 mg/mL 的奥司他韦对照品溶液。取奥司他韦原料药50 mg,精密称定,置50 mL 容量瓶中,加甲醇溶解并定容,摇匀,滤过,即得质量浓度为1 mg/mL 的奥司他韦供试品贮备液;量取上述贮备液2 mL,置10 mL 容量瓶中,用水稀释并定容,摇匀,即得奥司他韦供试品溶液。
2.3 奥司他韦含量测定
2.3.1 色谱分析
取2.2 项下各溶液,按拟订色谱条件进样分析。结果奥司他韦相对保留时间(RT)为4.18 min,峰形良好。色谱图见图1。
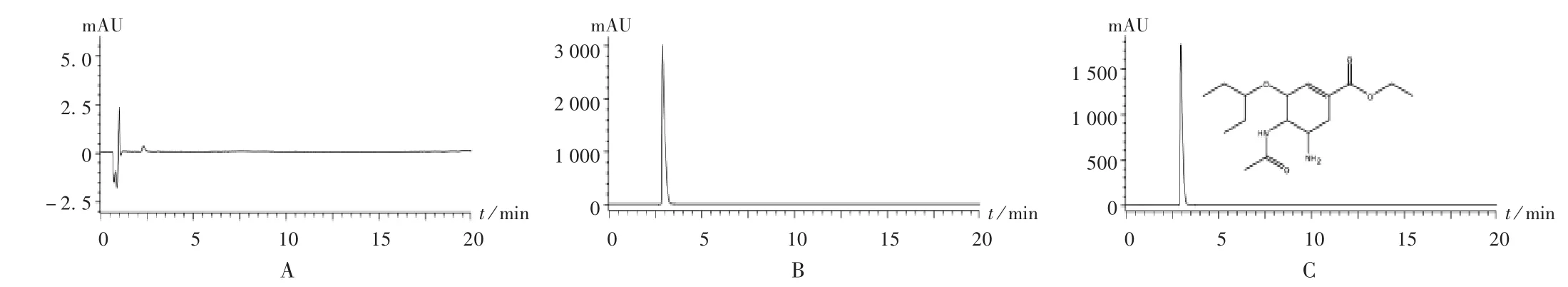
图1 奥司他韦高效液相色谱图
2.3.2 奥司他韦裂解规律
在UPLC-Q-TOF-MS 系统中,将奥司他韦对照品溶液以流动注射方式进样(流速为0.1 mL/min),获得一级(MS1)和二级(MS2)质谱图。奥司他韦分子式为C16H28N2O4,单同位素精确质量为312.204 9。MS1 中m/ z 313.212 8 为[M+H]+(见图2 A),与理论值m/ z 313.212 2 比较,误差为-1.91×10-6,表明仪器精密度高,结果准确、可靠。
对奥司他韦二级质谱图进行解析(见图2 B),其裂解碎片规律主要根据准确分子方程式,等效双键数(DBE),奇偶数互变脱氮,由分子结构特性等进行推测,其主要碎片包括m/ z 226.107 7,208.097 1,166.086 3,138.054 4,120.044 4。

图2 奥司他韦标样质谱图
奥司他韦的加氢离子[M+H]+的m/ z 为313.212 8,分子结构式为C16H29N2O4+,DBE 为4.0。碎片离子m/ z 226.107 7 的精确离子结构式为C11H16NO4+,DBE 为4.0。由于m / z 在碎裂前后由奇数变为偶数,表明有N 的脱落,而DBE 没有发生改变,且没有O 的变化,表明脱落的N 为连接在环上的氨基。根据分子结构,脱落的C5H13为醚上的碳支链。碎片离子m/ z 208.097 1,其精确离子结构式为C11H14NO3+,DBE 为5.0,在碎片离子m/ z 226.107 7 的基础下脱去1 个H2O,根据结构及DBE 值增加1 分析,脱去位置为环上的酚羟基,并形成双键。
奥司他韦有关物质提取离子流图见图3。由碎片离子m/ z 208.097 1 可见,可发生多个途径的分子碎裂:1)碎片离子 m/ z 166.086 3,其精确离子结构式为C9H12NO2+,DBE 为4.0,在碎片离子m/ z 208.097 1 的基础上脱去C2H2O,DBE 值减少1,因此减去了1 个含氧双键,但这个含氧双键不会是羧基上C=O,否则其后面的C2H5O 都会脱去,因此脱去的为连接在氨基上的羰基;2)碎片离子m/ z 138.054 4,其精确离子结构式为C7H8NO2+,DBE 为4.0,在碎片离子m/ z 208.097 1 的基础下脱去C2H2O+C2H4,因此除了脱去连接在氨基上的羰基还脱去了酯上的乙基;3)碎片离子m/ z 120.044 4,其精确离子结构式为C7H6NO+,DBE 为4.0,在碎片离子m/ z 138.054 4 基础上继续脱去1 个H2O,只能发生在羧基上脱水。奥司他韦分子的裂解规律分析与文献[7-9]报道一致,有助于分析奥司他韦降解规律,以及分解产物和杂质结构的鉴定。
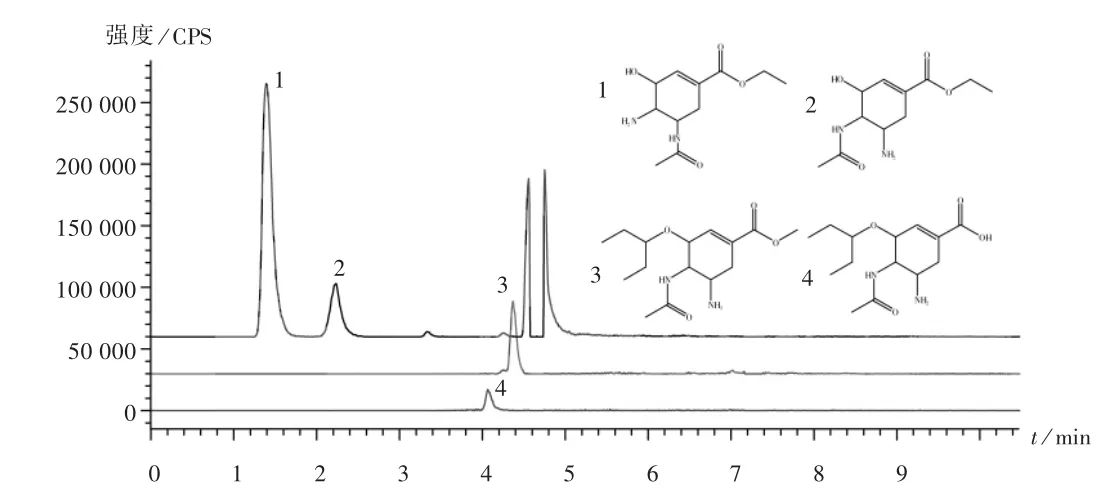
图3 奥司他韦有关物质提取离子流图
2.4 奥司他韦有关物质检测及结构鉴定
在奥司他韦原料药中共鉴定出4 种有关物质(杂质1,2,3,4),其保留时间分别为1.41,2.25,4.36,4.08 min,详见图3。杂质3 和杂质4 为2015 年版《中国药典(二部)》磷酸奥司他韦收录杂质。杂质1 和杂质2 尚未见报道,可能为药品合成工艺中的副产物,也可能是由于奥司他韦药物长期储存发生分解产生,其安全性未知。
杂质1 出峰时间为1.41 min,其一级质谱图中m/ z 243.133 8 为[M+H]+(见图4 A),分子式预测结果为C11H18N2O4,与理论值m/ z243.133 9 误差为-0.59×10-6。按和奥司他韦裂解规律类似的方法推导杂质的结构,其可能的结构见图3。二级质谱主要碎片为m/ z 208.096 4,166.085 7,137.070 1,120.043 8,109.075 6(见图4 B),可能的二级裂解规律推导见图5 A。
杂质2 出峰时间为2.25 min,其一级质谱图中m/ z 243.133 6 为[M+H]+(见图4 C)。分子式预测结果为C11H18N2O4,与理论值m/ z243.133 9 误差为-1.40×10-6。由于杂质2 的出峰时间在杂质1 后,推测其可能的结构见图3。二级质谱主要碎片为m/ z 166.085 7,162.052 6,137.070 1,120.043 8,109.075 6(详见图4 D),可能的二级裂解规律推导见图5 B。
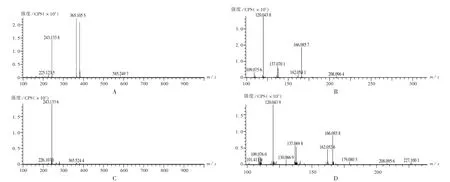
图4 奥司他韦杂质1 和杂质2 的一级和二级质谱图
2.5 方法学考察
专属性试验:称取奥司他韦原料药50 mg,精密称定,置50 mL 容量瓶中,按拟订色谱条件进样分析,记录色谱图。结果主峰与以上破坏产生的降解物峰均能获得良好分离,不干扰奥司他韦的测定。
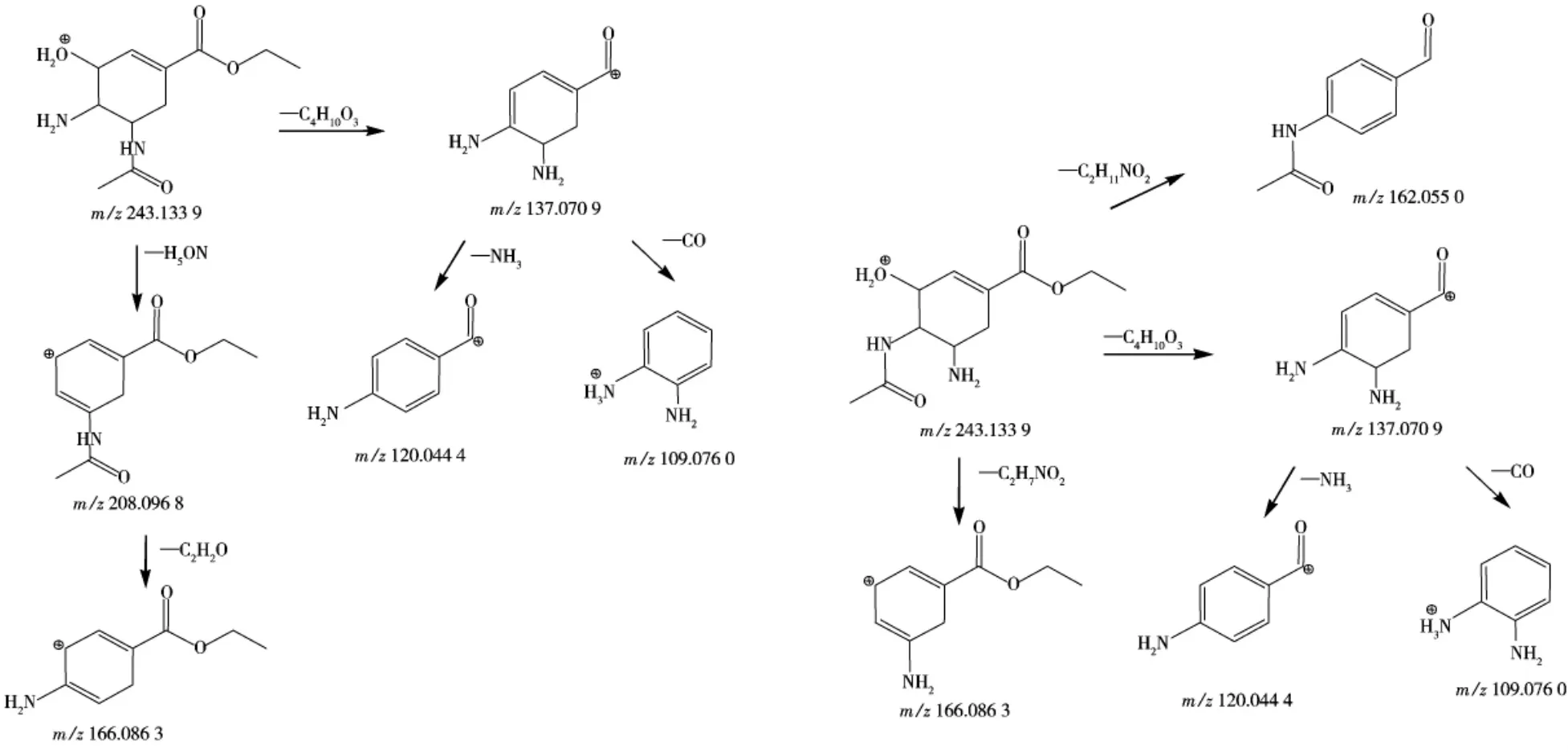
图5 奥司他韦杂质1 和杂质2 的二级碎片裂解推导图
线性关系考察:取奥司他韦对照品适量,精密称定,加水溶解并稀释成质量浓度为0.25,0.5,1.0,5.0,12.5 μg/mL 的溶液,按拟订色谱条件进样20 μL,测定。以峰面积(A)为纵坐标、质量浓度(C)为横坐标进行线性回归,得回归方程A=24.15C+1.213,r=0.9996(n=5)。结果表明,奥司他韦质量浓度在0.25~12.50 μg/mL范围内与峰面积线性关系良好。
定量限和检测限:取奥司他韦对照品溶液,逐步稀释,按拟订色谱条件进样测定。以信噪比(S/ N)=10 计,奥司他韦定量限为9 ng;以S/ N=3 计,检测限为3 ng。
精密度试验:取奥司他韦对照品溶液,按拟订色谱条件声进样测定,重复进样5 次。结果的 RSD 为0.17%( n=5),表明仪器精密度良好。
3 讨论
采用UPLC-Q-TOF-MS 法对奥司他韦药物的结构及其分子在质谱中的裂解规律进行精确地定性分析,其裂解碎片的质量数与理论值间的偏差均在10-6量级,证明了方法的准确性及结构推导的可靠性。本研究中对奥司他韦碎片裂解的定性分析可以用于奥司他韦药物的质谱快速鉴定,以及食品、环境等各种基质中的残留检测及药物评价。
本研究中首次鉴定出奥司他韦原料药中2 种新型有关物质。有可能为药品合成工艺中的副产物,也可能是由于奥司他韦药物长期储存发生分解产生,其服用后对生物体的影响未知,其药用效果及毒理有待后续评价。
