一种五元羧酸与Eu(Ⅲ)、Tb(Ⅲ)配合物的结构及光致发光性质
2020-09-10何盼盼王宏胜
王 磊 何盼盼 王宏胜
(许昌学院化学化工学院,许昌 461000)
0 引言
稀土配位聚合物的合成与性质研究是当前稀土配位化学研究的热点领域之一,这类配合物在光致发光、电致发光、荧光探针、磁性、吸附、催化等材料领域具有潜在的应用价值[1-6]。铕和铽在稀土配合物发光材料方面是应用最多的2种元素,一方面这2种元素三价离子的发光处于可见光区,分别能发射三基色中的红色和绿色光;另一方面,使用它们作为配位中心离子时,理论上可得到内量子发光效率非常高的发光材料[7-9]。利用含有共轭结构的有机配体与这2种稀土离子合成配合物是制备发光配合物的重要途径,当稀土离子的激发态能级与配体三重态能级的能级匹配,能量传递效率较高时,就可能制备出具有良好发光性能的发光配合物。另外,也可以通过同时使用2种含共轭结构的有机配体的方法提高配合物的发光性能,比如一种配体使用芳香有机羧酸,另一种配体用1,10-菲咯啉、2,2′-联吡啶等[10-13]。不论选择几种配体,配体的三重态能级与稀土离子激发态能级的匹配对发光效率的影响都是非常重要的。因此,选择合适的配体对于稀土配合物的发光性能起着主要的作用。目前,已有大量含共轭结构的有机羧酸与三价稀土铕和铽离子反应合成发光配合物方面的报道[14-16],也有一些研究者利用2种有机羧酸作配体来合成有新颖结构和多种性质的多功能稀土配合物[17-19]。本文中我们报道了一种含有共轭结构的新型五元有机羧酸:4-(4-羧基苯基)吡啶-2,3,5,6-四羧酸(H5L,Scheme 1),并利用此酸做配体,与稀土铕(Ⅲ)和铽(Ⅲ)离子反应,合成了2种配合物,测定了配合物的结构,并对其光致发光性能进行了研究。
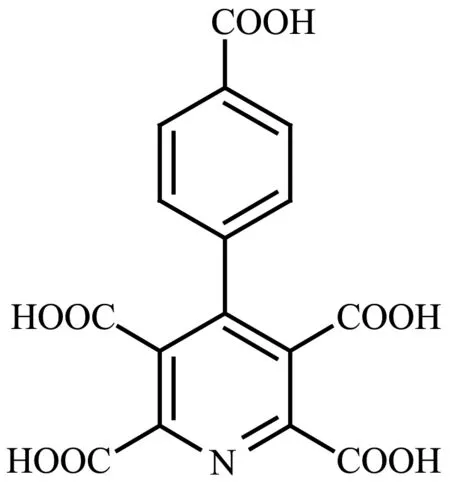
Scheme 1 Structure of ligand H5L
1 实验部分
1.1 试剂与仪器
Eu(ClO4)3溶液通过高氯酸(3 mol·L-1)和 Eu2O3(≥99.99%)反应制备。Tb(ClO4)3溶液通过高氯酸(3 mol·L-1)、Tb4O7(≥99.95%)及 H2O2(30%)的反应制备。2,6-二甲基-4-(4-甲基)苯基-3,5-吡啶二甲酸二乙酯根据文献方法合成[20]。实验用水为去离子水,其它试剂均为分析纯。
实验中配合物的荧光性质包括激发、发射光谱、量子产率和荧光寿命,使用爱丁堡仪器公司的FLS980稳态/瞬态荧光光谱仪进行测试。红外光谱用FTIR-650型红外光谱仪(KBr压片法)进行测试。熔点用X-4显微熔点仪测定。核磁共振光谱用布鲁克公司AV-400核磁共振波谱仪测试。
1.2 配体H5L的合成
在100 mL圆底烧瓶中加入5.8 g(17 mmol)2,6-二甲基-4-(4-甲基)苯基-3,5-吡啶二甲酸二乙酯、1.7 g氢氧化钠和30 mL水配成的溶液、20 mL工业乙醇,在磁力搅拌下回流反应12 h。旋蒸除去乙醇,补加20 mL水,分批加入12 g高锰酸钾,90℃下进行氧化反应。溶液褪色后,用浓盐酸调节pH值到1~2,析出大量白色固体,冷却,抽滤,干燥。用水进行重结晶,得到白色固体2.7 g(产率42.1%)。1H NMR(400 MHz,DMSO-d6):δ8.04(s,2H),7.97~7.95(m,2H),7.38~7.36(m,2H)。13C NMR(101 MHz,DMSO-d6):δ167.4,167.1,166.0,147.1,145.7,139.8,135.0,133.6,131.4,129.9,129.4,129.1。
1.3 配合物1和2的合成
称取0.038 0 g五元酸(0.1 mmol)放入反应釜内胆中,加入2.5 mL浓度为0.040 mol·L-1的高氯酸铕水溶液,加入10 mL去离子水,用NaOH溶液调节pH值为5。将反应釜放入烘箱中,在170℃反应72 h后,降温至室温,得到配合物的无色晶体,产率58%(基于Eu(Ⅲ)离子计算)。配合物2的制备方法与配合物1的相同,只是将高氯酸铕水溶液改为高氯酸铽水溶液,其产率为52%(基于Tb(Ⅲ)离子计算)。对配合物进行熔点测试表明,二者的熔点都在300℃以上。
1.4 配合物的结构表征
选取尺寸为0.20 mm×0.15 mm×0.11 mm的配合物1和0.26 mm×0.21 mm×0.15 mm的配合物2的单晶,在Bruker APEX-ⅡCCD型X射线单晶衍射仪上,用经石墨单色器单色化的MoKα射线(λ=0.071 073 nm),以φ-ω扫描方式在一定的θ范围内收集衍射数据。利用SADABS程序对数据进行吸收校正,晶体结构通过直接法用SHELXS-97[21]程序进行初步解析,并用SHELXL-97[22]程序对粗结构进行精修,精修参数列于表1。部分非氢原子通过差值Fourier合成确定,对非氢原子的坐标及各向异性热参数进行了全矩阵最小二乘法修正;非水分子上的氢原子通过理论加氢确定。2个配合物晶体结构中的水分子存在严重的位置无序,故这些水分子上的氢原子坐标未能精确确定,因此在结构解析中水分子上的氢原子没有添加[23]。2种配合物中的Eu(Ⅲ)和Tb(Ⅲ)离子与氧原子或氮原子的键长值、部分键角值分别列于表S1(Supporting information)和表S2中。
CCDC:1949202,1;1949204,2。
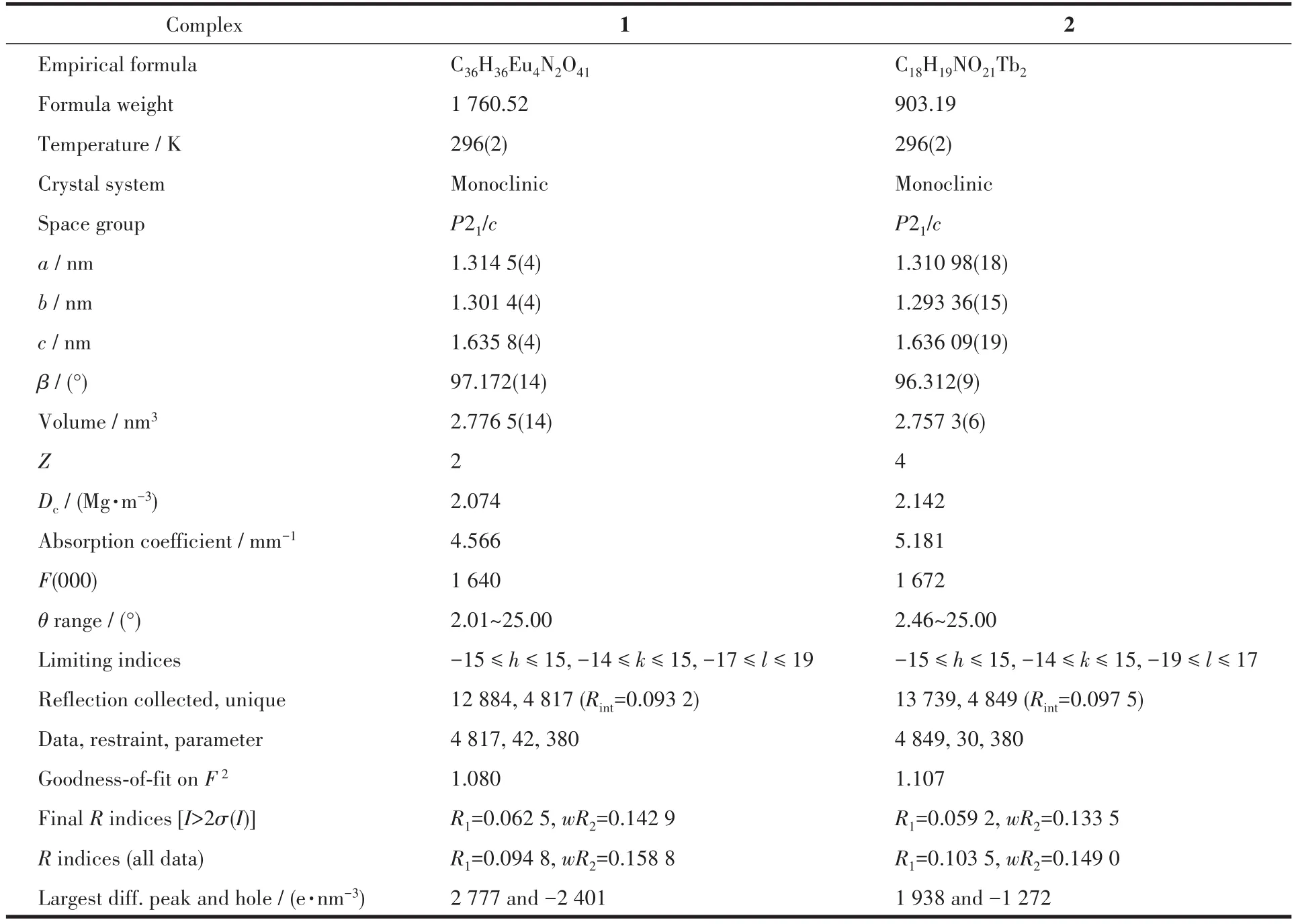
表1 配合物1和2的晶体学数据Table 1 Crystallographic data for complexes 1 and 2
2 结果与讨论
2.1 配合物的结构
由表1可知,配合物1和2的化学式分别为{[Eu4(HL)2(ox)2(H2O)10]·3H2O}n和 {[Tb2(HL)(ox)(H2O)5]·2H2O}n,虽然稍有不同,但是可以看出这种差异是由于晶格水的数目不同导致的结果。其晶系和空间群完全相同,所以,这2个配合物属于同构化合物。在合成过程中,没有加入草酸或其盐类化合物,故配合物中的草酸根离子可能是在水热条件下,部分配体发生分解产生。测试二者的红外光谱,可以看出谱图非常相似,表明二者具有相似的结构,这与单晶结构测试的结果相一致,2种配合物的红外光谱图见图S1。以配合物1为例来描述这2种配合物的结构。配合物1中铕离子有2种配位形式,二者的配位数都是8(图1),其中Eu1与1个N原子和7个氧原子配位,氧原子中有2个来自草酸根离子,其余5个来自4个五元羧酸配体。8个配位原子形成了变形双帽三棱柱多面体结构,Eu1位于多面体内部,如图2a所示。Eu2的8个配位原子都是氧原子,其中2个同样来自草酸根离子,其余6个中5个来自配位水分子,1个来自五元羧酸配体的羧基。这8个配位原子形成的配位多面体与Eu1的配位原子形成的多面体不同,其更接近变形的四方反棱柱结构,如图2b所示。与Eu1配位形成的8个配位键分别为Eu-O1、Eu-O3、Eu-O5、Eu-O8、Eu-O9、Eu-O11、Eu-O13和Eu-N1,其对应的键长分别为0.235 2(7)、0.234 7(9)、0.233 8(7)、0.230 6(10)、0.238 7(9)、0.245 7(9)、0.243 4(10)和0.251 2(10)nm。与Eu2配位形成的8个配位键分别为 Eu-O1w、Eu-O2w、Eu-O3w、Eu-O4w、Eu-O5w、Eu-O2、Eu-O12及Eu-O14,其对应的键长分别为 0.236 8(16)、0.24 1(3)、0.237(2)、0.244(2)、0.243 8(15)、0.246 9(8)、0.239 5(9)和 0.240 4(10)nm。以Eu1为顶点的O-Eu-O键角最大为148.7(3)°,最小为65.9(3)°;O-Eu-N键角的最大值为141.5(3)°,最小值为63.6(3)°。以Eu2为顶点的O-Eu-O键角最大值为147.6(6)°,最小值为65.1(9)°。
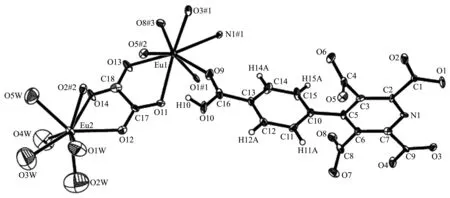
图1 配合物1中Eu(Ⅲ)离子的2种配位环境Fig.1 ORTEP representation of 1 showing two coordination environments of Eu(Ⅲ)
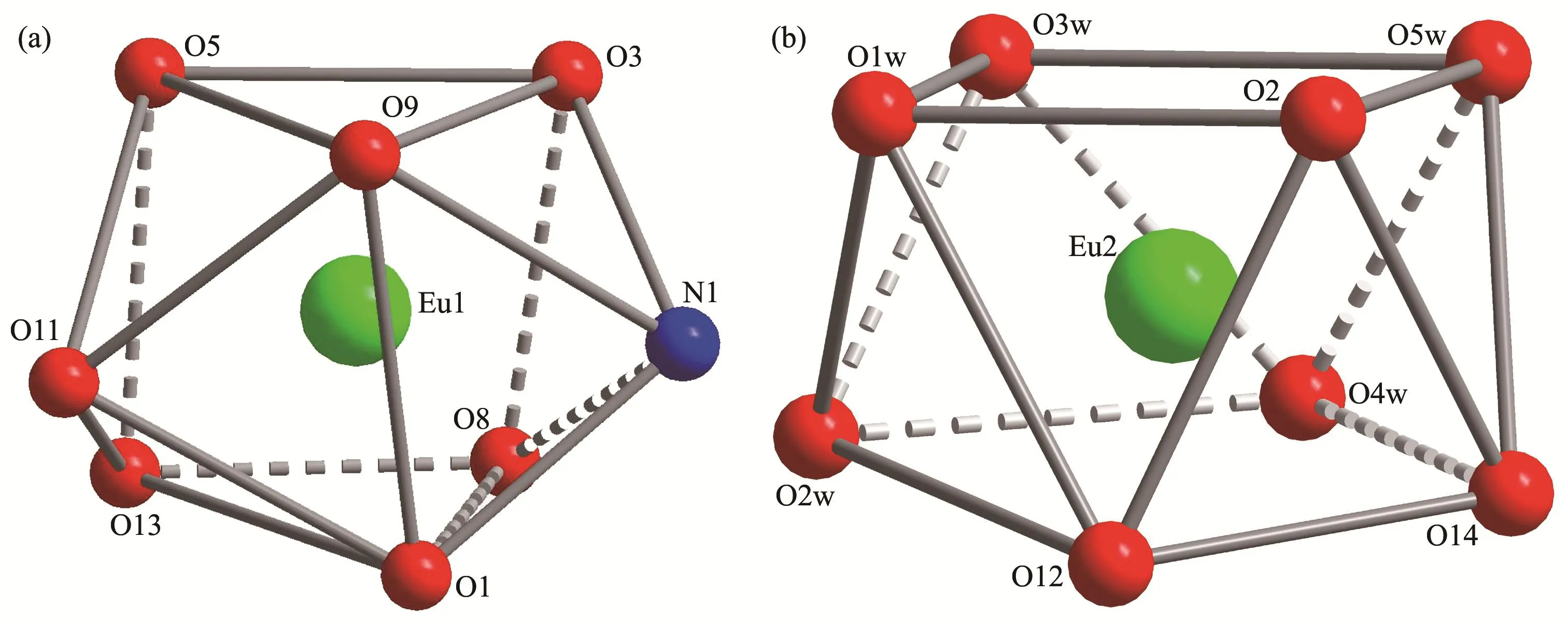
图2 Eu1和Eu2分别与8个配位原子形成的变形双帽三棱柱(a)和四方反棱柱多面体(b)Fig.2 Distorted bicapped trigonal prism(a)and square antiprism(b)formed by eight coordination atoms with Eu1 and Eu2,respectively
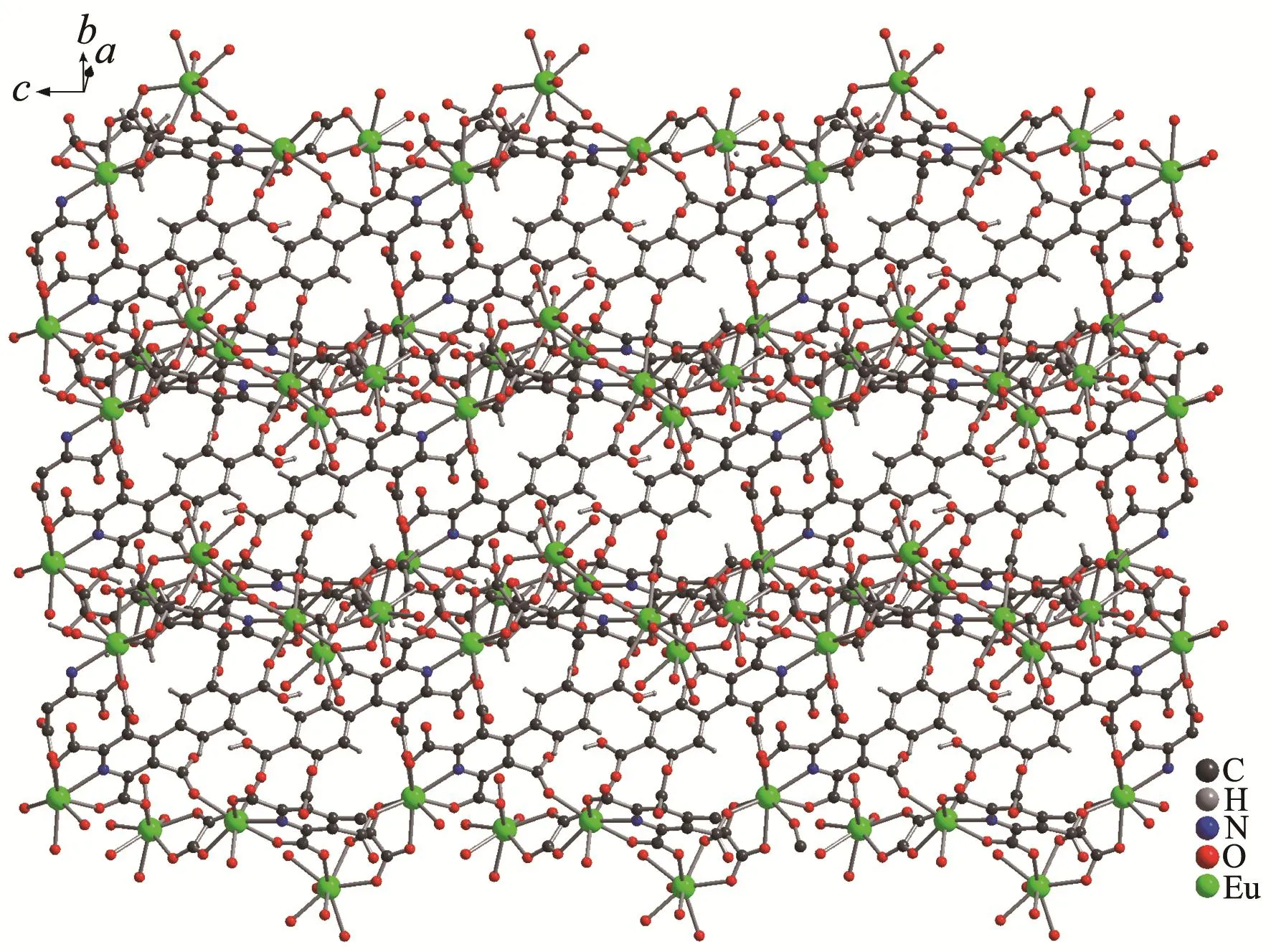
图3 配合物1中由五元酸及草酸与Eu(Ⅲ)离子形成的二维结构Fig.3 Two-dimensional structure formed by pentacarboxylate,oxalate ligands and Eu(Ⅲ)for 1
五元羧酸配体在配合物中失去4个氢离子,以四价阴离子的形式配位,其中吡啶环上的4个羧基均失去氢离子以羧酸根的形式与Eu(Ⅲ)离子配位,而苯环上的1个羧基并没有失去氢离子,是以羧基上的羰基氧原子O9与Eu(Ⅲ)离子配位(图1)。每个五元羧酸配体连接了5个Eu(Ⅲ)离子,其中,吡啶环上的2-、6-位羧基与N原子以三齿螯合方式与1个Eu(Ⅲ)离子配位;同时,2-位羧基的另一个氧原子以单齿配位方式还连接了1个Eu(Ⅲ)离子;3-、5-位上的羧基均以单齿配位形式各连接1个Eu(Ⅲ)离子;苯环上的1个羧基也以单齿配位方式连接1个Eu(Ⅲ)离子。
草酸根离子以螯合配位方式连接了2个Eu(Ⅲ)离子,这种配位方式与文献报道的配位方式一致[24-26]。通过草酸根离子和五元羧酸配体与Eu(Ⅲ)离子的配位,生成了一个二维平面结构的配位聚合物,如图3所示。
2.2 配合物的光致发光性质
以发射波长611 nm作为监测波长,在200~450 nm波长范围测试配合物1的粉末样品的激发光谱,得到的结果如图4a所示,其最佳激发波长为300 nm。以此波长为激发波长,测试配合物1的发射光谱,结果如图4b所示,在580、591、611、650和699 nm处出现了5个发射峰,分别对应于Eu(Ⅲ)离子的5D0→7FJ(J=0,1,2,3,4)的跃迁。其中的5D0→7F2的跃迁为Eu(Ⅲ)离子电偶极跃迁,5D0→7F1的跃迁为Eu(Ⅲ)离子的磁偶极跃迁,电偶极跃迁强度为磁偶极跃迁强度的4倍多,表明Eu(Ⅲ)离子在配合物中不处于反演中心的位置上[27]。
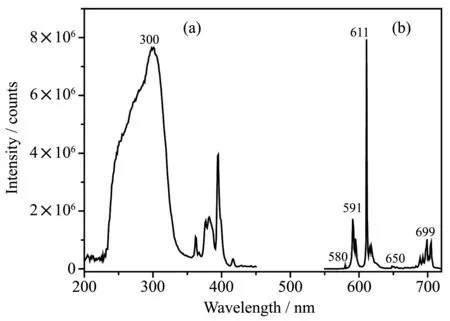
图4 配合物1的激发光谱(a)和发射光谱(b)Fig.4 Excitation spectra(a)and emission spectra(b)of complex 1
以发射波长545 nm作为监测波长,在200~425 nm波长范围内测试配合物2的粉末样品的激发光谱,得到如图5a所示的较宽的激发峰,其最佳激发波长为338 nm。以此波长为激发波长,测定配合物2的发射光谱,结果如图5b所示,可以看到在489、545、584和620 nm处有4个很强的发射峰,分别对应于Tb(Ⅲ)离子的5D4→7FJ(J=6,5,4,3)跃迁。除此之外,在图中也可以看到在650、667和680 nm处有3个较弱的发射峰,其强度明显比前4个峰弱很多,这3个峰分别对应于 Tb(Ⅲ)离子的5D4→7FJ(J=2,1,0)跃迁。因此,该配合物的发射光谱中,可以看到Tb(Ⅲ)离子5D4能级向基态跃迁时产生的全部7个发射峰,同时也表明该配合物具有较好的发光性能。
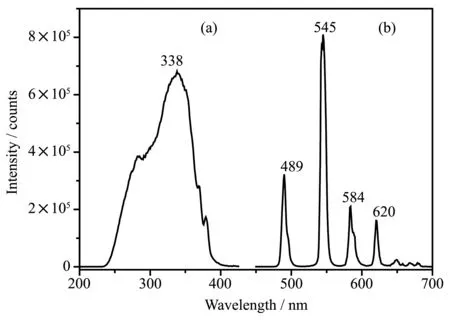
图5 配合物2的激发光谱(a)和发射光谱(b)Fig.5 Excitation spectra(a)and emission spectra(b)of complex 2
设定激发波长和发射波长均为300 nm,用积分球测定配合物1粉末样品的发射光谱,通过仪器软件进行数据处理得出配合物1的绝对量子产率为45%。用同样的方法,设定激发波长和发射波长均为338 nm,测得配合物2的绝对量子产率为38%。从这2个配合物的绝对量子产率也可以看出,二者都具有较好的发光性能。
2.3 配合物的荧光寿命
设定激发波长和发射波长分别为300和611 nm,以微秒脉冲氙灯为激发光源,调节合适的激发和发射狭缝,测定配合物1中Eu3+的5D0激发态的衰减曲线,并进行非线性拟合,衰减曲线和拟合曲线见图6。拟合公式为y=A1exp(-x/t1)+y0,拟合后得到配合物1的5D0激发态寿命为1.83 ms,R2为0.998,A1为2 870.14,y0为15.71。同样,设定激发波长和发射波长分别为338和545 nm,测试配合物2的5D4激发态的寿命,其衰减曲线和拟合曲线如图7所示。所采用的拟合公式同上,其R2的值为0.997,A1为2 648.35,y0为 22.82,测得的配合物 2的5D4激发态的寿命为1.07 ms。
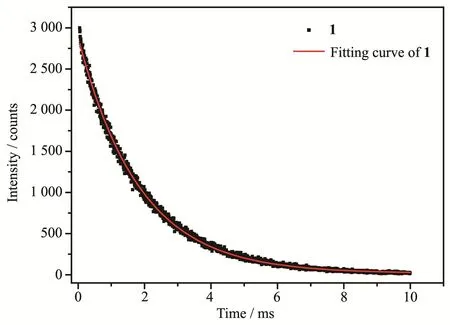
图6 配合物1的荧光衰减曲线及拟合曲线Fig.6 Decay curve and fitting curve of complex 1
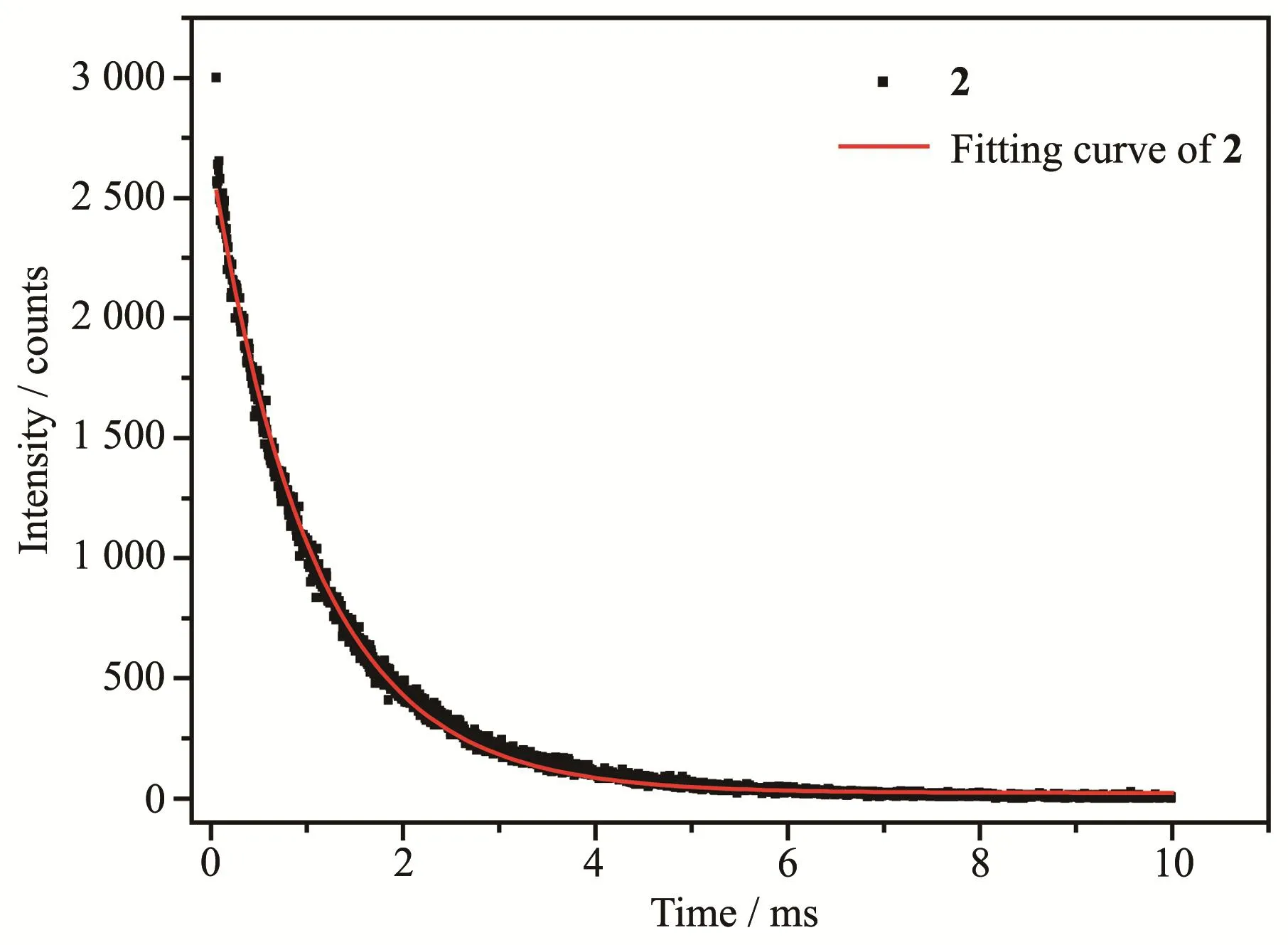
图7 配合物2的荧光衰减曲线及拟合曲线Fig.7 Decay curve and fitting curve of complex 2
3 结 论
采用水热合成方法,用高氯酸铕和高氯酸铽分别与一种五元羧酸(4-(4-羧基苯基)吡啶-2,3,5,6-四羧酸)反应,合成了稀土铕和铽的配合物。反应过程中部分五元羧酸发生分解,产生了草酸,所以形成的配合物中含有草酸根离子配体。这2个配合物结构相同,为2D层状结构。二者在紫外光的激发下,均可发射很强的荧光,其中铕配合物的绝对发光量子产率为45%,铽配合物的绝对发光量子产率为38%。
Supporting information is available at http://www.wjhxxb.cn
