HPLC-MS/MS测定豆类及其制品中的赭曲霉毒素A含量
2020-09-07葛晓晓彭雪琦江苏省理化测试中心
□ 葛晓晓 彭雪琦 江苏省理化测试中心
赭曲霉毒素(ochratoxin)是一种天然存在的毒性污染物质,有A、B、C与D 4种,其中赭曲霉毒素A(ochratoxin A,简称OTA)毒性最大[1]。赭曲霉毒素广泛存在于粮食、豆类、咖啡豆和饲料等,在存储及运输方式不当时,食品发生霉变可产生。OTA对动物及人类的免疫系统有显著性的破坏作用,对动物及人类肾脏、肝脏等具有致癌、致畸等作用[2—3],所以食品中OTA含量检测至关重要。
目前,我国对于豆类及其制品中赭曲霉毒素A的定量检测方法主要包括液相色谱(HPLC)[4]和液相色谱串联质谱(LC—MS/MS)法[5]。HPLC 法采用荧光检测器检测,准确性高。LC—MS/MS法具有更高的灵敏度和高效性,但实验前处理方法对检测结果的干扰较大。常用的净化方法有免疫亲和柱净化[6]、离子交换固相萃取柱净化[7]、酶联免疫吸附法[8]等。此外,目前文献中对于豆类及其制品中赭曲霉毒素A测定技术的研究较少。本研究基于LC—MS/MS检测技术,在现有检测方法基础上对豆类及其制品的赭曲霉毒素A提取及测定方法进行优化,研究成果可为实验室检测赭曲霉毒素A提供可靠依据。
1 实验部分
1.1 仪器、试剂与材料
Agilent1290—6470 液相色谱 —三重四级杆串联质谱仪(美国,安捷伦公司);BAS224S—CW万分之一天平(赛多利斯);KQ—400KDE高功率数控超声波清洗器(昆山市超声仪器有限公司);TGL—18高速离心机(上海安亭科学仪器厂);RE—2000B旋转蒸发仪(上海亚荣生化仪器厂);SHB—IIIA循环水式多用真空泵(上海豫康科教仪器设备有限公司);AutoVapS60氮吹浓缩仪(美国ATR)。
赭曲霉毒素 A 标准品(10 μg/L,Pribolab Pte Ltd),氯化钠,甲醇(AR级,国药集团);甲醇,乙腈,甲酸(HPLC级,美国Honeywell公司);免疫亲和柱(Romer Labs)。
1.2 仪器工作条件
1.2.1 液相色谱条件
色谱柱Waters Sunfire C18(100 mm×2.1 mm,3.5 μm); 色 谱 柱 温 度40 ℃;进样量 20 μL。流动相 A 为0.1%甲酸,流动相B为乙腈,梯度洗脱:0~0.20 min(80% A+20% B),0.20 ~ 0.50 min(30%A+70%B),0.80 ~ 0.90 min(80%A+20%B),0.90~2.50 min(80%A+20%B),流速0.5 mL/min。
1.2.2 质谱条件
质谱扫描采集方法MRM,质谱离子源为ESI电离源,采用正离子模式,其他质谱条件如表1所示。

表1 质谱参数
1.3 实验方法
1.3.1 样品前处理方法
样品粉碎,过筛,称取25 g(精确到0.001 g)于100 mL容量瓶中,加入5 g氯化钠,加入60%乙腈溶液稀释定容,超声20 min,取部分溶液过滤,离心,取上清液。精密量取滤液10 mL于离心管中,加入40 mL水混匀,取10 mL过0.22 μm滤膜过免疫亲和柱,用5 mL甲醇洗脱,氮气吹干,加入1 mL甲醇溶解,待测。
1.3.2 标准曲线配制
溶剂标准溶液:准确移取赭曲霉毒素A标准溶液1 mL于10 mL容量瓶,稀释定容。分别移取5、10、20、50、100 μL 与 200 μL, 于 6 个10 mL容量瓶,用甲醇溶液稀释定容,配制成浓度为0.5、1、2、5、10 μg/L和20 μg/L的系列标准溶液,待测。
2 结果与讨论
2.1 前处理优化
本次检测参考GB 5009.96—2016,对萃取溶剂进行了优化,研究了甲醇、乙腈、80%甲醇、80%乙腈、60%乙腈的提取效果。结果表明,加入少量水有助于回收率的提高,但水过多,会使提取过程中混入更多植物蛋白等水溶性物质。因此结合文献[9]及标准[10],后续试验选择60%乙腈为提取溶剂。
2.2 色谱条件优化
试验考察了Waters Sunf i re C18(100 mm×2.1 mm,3.5 μm),Agilent Poroshell 120 EC—C18(50 mm×3.0 mm,2.7 μm)两种不同色谱柱对赭曲霉毒素A的测定效果。结果表明,在同等条件下,采用C18短柱,响应值更高,所以采用C18短柱。试验分别采用甲醇—水,乙腈—水作为流动相,发现赭曲霉毒素A峰形较宽。由于赭曲霉毒素 A 含有氨基(—NH2)及氯(—Cl),所以在水相中加入0.1%甲酸,可促进离子化效应,以提高响应。采用1%甲酸—甲醇作为流动相时,甲醇出峰时间更早,不利于杂质与目标物质的分离。所以试验采用0.1%甲酸水—乙腈作为流动相,分离效果好。
2.3 方法学研究
取阴性空白样,按相同前处理方法制得其空白基质溶液,配制20 μg/L的基质标准溶液,临用现配。同时用甲醇配制相同浓度的标准溶液,上机平行测定6次。实验结果显示,基质标准溶液峰面积为甲醇标准溶液峰面积的108%,说明经过前处理后,净化程度较高,基质效应影响较小。在后续试验中,可采用甲醇配制标准溶液。
试验采用甲醇配制质量浓度为0.5~20 μg/L的赭曲霉毒素A标准系列溶液。按照优化后的色谱、质谱条件依次进行测定。对质量浓度(x,μg/L)和定量离子对峰面积(y)进行线性拟合。拟合结果表明在0.5~20 μg/L范围内线性关系良好,线性方程为y=14 073.8x—902.9(R2=0.999 4)。 采用标准添加法,以信噪比S/N≥10确定定量限(LOQ)为1 μg/kg,逐级稀释,以信噪比S/N≥3确定检出限(LOD)为 0.3 μg/kg。GB 2761—2017[11]规 定豆类及其制品中赭曲霉毒素A的限量为5.0 μg/kg,所以定量限满足检测需求。
选择多种阴性样品进行添加回收率和精密度试验,试验随机选择了3种食品—黄豆、豆干、绿豆。添加水平分别为1、2、10 μg/kg,按本文方法进行样品前处理和测定,平行测定6次。结果见表2。由表2可见,在表中3种食品中,赭曲霉毒素A的平均回收率为85.6%~106.2%,相对标准偏差(RSD)为0.96%~3.5%,符合检测要求。
2.4 样品检测
随机抽检20份豆类及其制品食品,采用本文建立的方法测定其中赭曲霉毒素A的含量,均为阴性。采用GB 5009.96—2016第一法复核,结果一致。说明本文方法可行。
3 结论
本文建立了一种基于高效液相色谱—串联质谱检测技术的豆类及其制品中赭曲霉毒素A含量的检测方法,该方法以80%甲醇提取结合免疫亲和柱净化,前处理过程便捷、快速,且检测结果精确,该方法可为检测豆类及其制品提供新的选择。
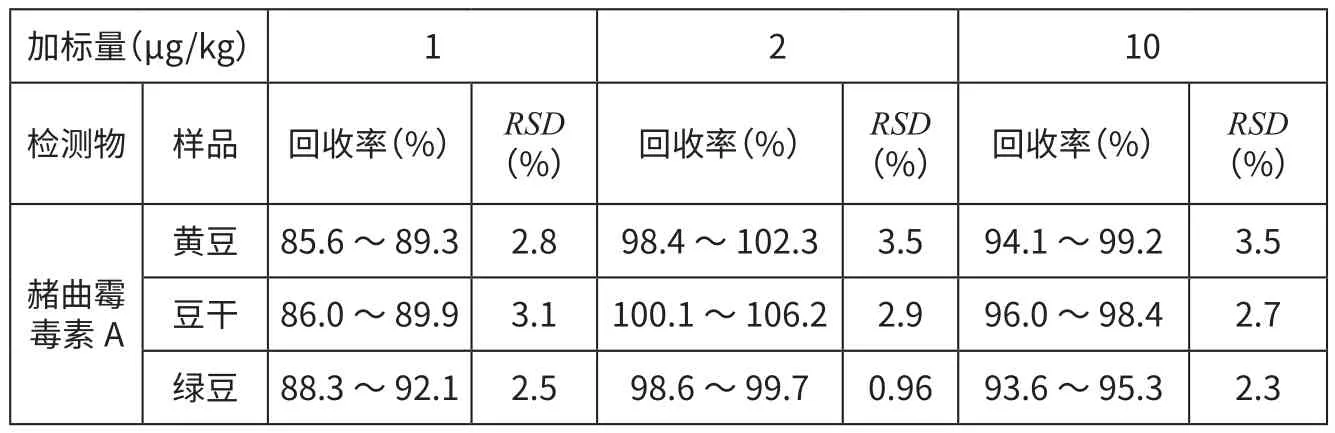
表2 赭曲霉毒素A的加标回收率和精密度(n=6)
