水分子在伊利石表面的吸附作用机理分析
2020-09-03邱鸿鑫陈浙锐王光辉
邱鸿鑫,陈浙锐,王光辉
中国矿业大学化工学院,江苏 徐州 221116
伊利石是影响煤炭浮选效果的主要粘土型矿物,在水中极易泥化,形成大量的带有负电荷的粘土颗粒,且具有强亲水性,易吸附水分子,在颗粒表面形成水化层,降低浮选效率,消耗大量浮选药剂,因此,研究伊利石与水分子的吸附具有重要意义。
随着分子动力学与量子化学的兴起,当前在探究矿物水化吸附机理上被广泛应用。王进等[1-3]通过动力学模拟分析了钠蒙脱石的水化膨胀与层间结构特征;Wang 等[4]应用分子动力学模拟云母表面水化膜的形成;Kerisit 等[5]通过分子动力学模拟了正长石的水化过程,得到了扩散系数的变化规律;陈攀等[6]研究了季铵盐在高岭石(001)面的吸附;Alvin 等[7]使用第一性原理研究了蒙脱石(001)面,发现含有的羟基对水分子吸附有着明显作用;彭陈亮等[8-9]研究了水分子在蒙脱石表面的吸附,解释了吸附主要是由静电相互作用产生;Long 等[10-11]使用DFT 模拟了水分子对黄药在闪锌矿与方铅矿上吸附位点的影响;韩永华等[12-13]研究了羟基钙离子对硅酸钠药剂在高岭石表面的吸附影响,确定了其吸附机理;何满潮、方志杰等[14-15]通过掺杂Fe/Mg/Al 等对蒙脱石的晶胞进行了研究;Bains 等[16]对粘土矿物水化膨胀进行了分子动力学模拟;Zhu 等[17]对TCDD 在蒙脱石上的吸附进行了动力学模拟;Zhao 等[18]利用量子化学模拟了Cu、Pb、Ni 和Hg 在高岭石表面的吸附构型;Michalkova 等[19]研究了有机小分子在高岭石表面的吸附构型。
为了解释水分子在伊利石表面吸附机理,本文选择从微观层面,使用Materials Studio 软件,预测了水分子的初始吸附位点,确定了稳定吸附构型,可视化的展示了水分子吸附位置,研究了水分子吸附前后的伊利石表面分子静电势变化,通过态密度与布居分析说明了水分子的吸附机理。
1 模型选择优化与计算
1.1 模型优化
伊利石是一种类似于云母的层状结构的粘土矿物,属于单斜晶系的硅酸盐矿物,文章选择由Drits 与Zviagina 构建的伊利石原胞模型[20],呈现2:1 型结构单元层的二八面体型,其中上下层均是铝、氧和硅形成的多面体,中间层是由钾离子填充,以补充晶胞中的阳离子,沿Z 轴方向的晶胞单元,含有Al-O-Si-O-K-O-Si-O-Al 共9 层原子,价电子分别是Al 3s23p1、O 2s22p4、Si 323p2、K 4s1。
通过Materials Studio 软件中的CASTEP 模块对原胞和水分子(水分子置入 15×15×15×15×10-3nm的晶胞中)进行几何优化,采用梯度泛函(GGA)的PBE 模块进行优化计算,使用Grimme 消除色散力影响,截断能选择340 eV,k 点选择Gamma,设置自洽场的收敛精度为 2×10-6eV/atom,设定几何优化标准:原子之间的最大作用力0.05 eV/A,原子之间的最大应力0.1 GPa,体系的能量变化 2×10-5eV/atom,优化后的结构见图1、2。
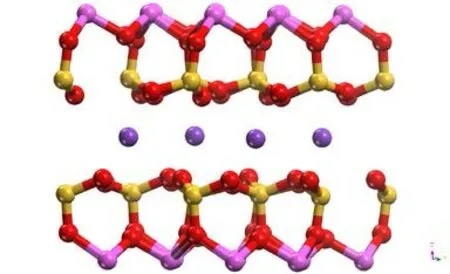
图 1 优化后伊利石主视Fig .1 Optimized illite main view
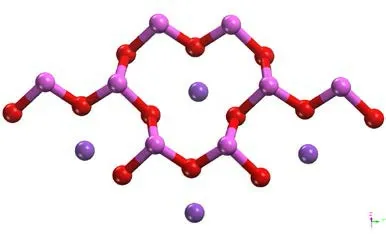
图 2 优化后伊利石俯视Fig .2 Optimized illite top view
对优化后的伊利石晶胞,使用Materials Studio软件中的Cleave Surface 模块对优化后的伊利石沿001 切分,切分面分为含有钾离子(IN-K-001)与不含钾离子(None-K-001)两个表面,为了消除周期性的边界影响,各添加3 nm 的真空层。
1.2 伊利石表面静电势
静电势适合用于分析由静电主导的弱相互作用,可以定性了解体系哪个部位更易发生静电相互作用。分子表面不同区域的静电势大小可以通过不同颜色来体现,分子之间易以静电势互补相结合,即一个分子表面静电势正值区域倾向于接触其他分子表面静电势负值区域,且数值反差越大,相互作用越强,这样能最大程度降低整体能量。
通过Multiwfn[21]软件可以计算出分子表面静电势的极大值点与极小值点的位置和数值,根据位置信息可以预测相互作用的位点,而数值可以判断静电相互作用强度,可以进行定量分析,其中青色代表极小值点,橘色代表极大值点。见图3、4。
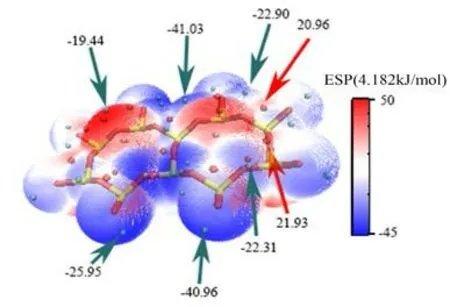
图 3 IN-K-001 面表面静电势Fig .3 IN-K-001 surface electrostatic potential
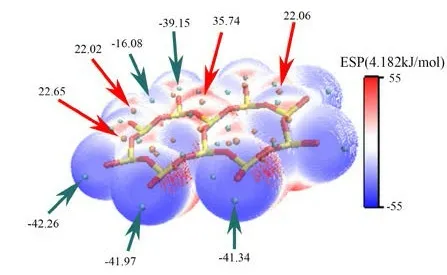
图 4 None-K-001 面表面静电势Fig .4 None-K-001surface electrostatic potential
1.3 水分子吸附位点预测
使用Adsorption Locator 模块对水分子在伊利石001(001)面的较佳吸附位点进行模拟计算,得到水分子初始吸附位点,见图5、6。
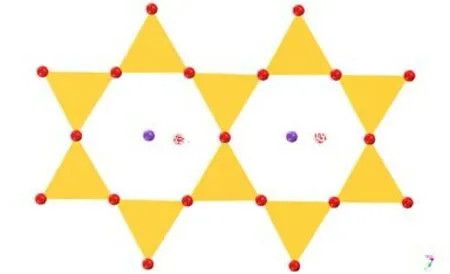
图 5 IN-K-001 面吸附位点俯视Fig . 5 Top view of the IN-K-001 surface adsorption site
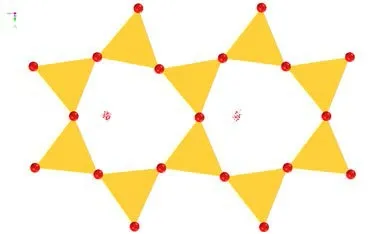
图 6 None-K-001 面吸附位点俯视Fig .6 Top view of the None-K-001surface adsorption site
2 结果分析
2.1 吸附能计算
当水分子在伊利石表面发生吸附作用时,整个体系将释放能量,即吸附能,计算公式如下:

其中, 代表水分子吸附到伊利石001 表面的吸附能, 为负值,说明反应可以自发进行,数值越小,反应越稳定,相反,若 为正,则反应无法正常进行, 代表水分子在伊利石表面稳定吸附时的体系总能量, 与 分别表示未发生吸附前,通过优化的水分子与伊利石表面的体系总能量。
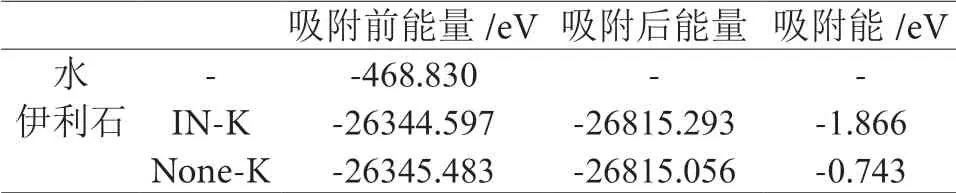
表1 水分子吸附于伊利石表面的吸附能Table 1 Adsorption energy of water molecules adsorbed on the surface of illite
见表1,水分子吸附在伊利石IN-K-001 面的吸附能为-1.866eV,对比水分子在伊利石None-K-001 面吸附能更低,说明水分子吸附于伊利石IN-K-001 面更稳定,说明伊利石解离后INK-001 面亲水性更强,水分子会优先吸附于伊利石IN-K-001 面。
2.2 吸附构型与Mulliken 布居分析
通过对预测的初始吸附模型的结构优化,得到水分子在伊利石IN-K-001 面与None-K-001 面上的稳定吸附构型,吸附结果见图7、图8。
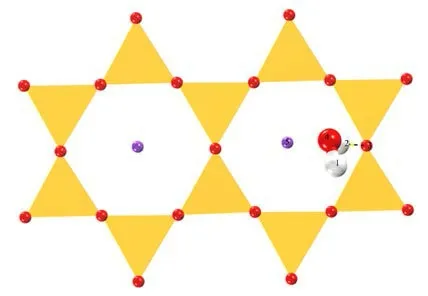
图 7 IN-K-001 面稳定吸附构型俯视Fig .7 Top view of the IN-K-001 surface stable adsorption configuration
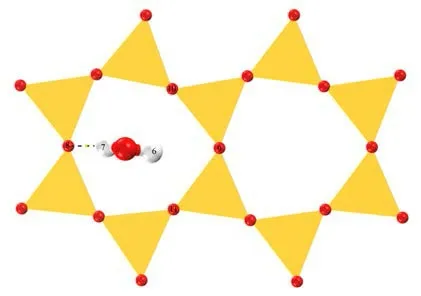
图 8 None-K-001 面稳定吸附构型俯视Fig .8 Top view of the None-K-001surface stable adsorption configuration
见图7,水分子垂直于伊利石表面,其中一个氢原子H2朝下,与伊利石IN-K-001 面一个氧原子O3形成了氢键。而氧原子O4虽然受钾离子K5静电吸引,却并未形成配位键,见图8,水分子中两个H 原子均朝下垂直于伊利石None-K-001 面,但是只有其中一个氢原子H7 与氧原子O8形成了氢键。
Mulliken 布居分析即通过对原子间的电荷进行平均划分,从而得到每一个原子的净原子布居和重叠布居,以此来作为原子间成键判断的一种方法依据,其取值范围为[0,1]。当电荷重叠的越多,则布居值越大,成键的共价性越强;当布居值为0时,则并未成键。
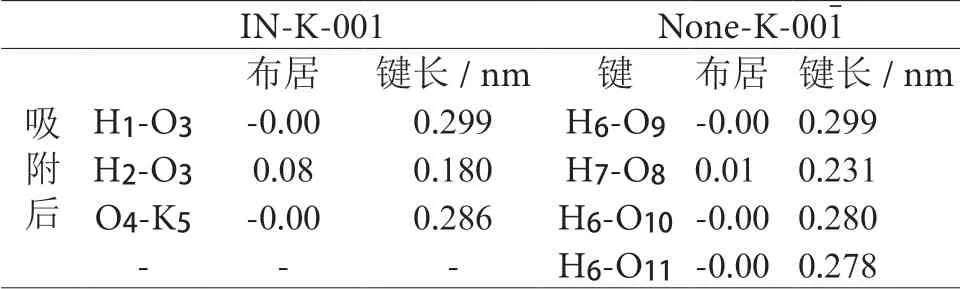
表 2 水分子吸附后的Mulliken 布居分析Table 2 Mulliken population analysis after adsorption of water molecules
见表2,在IN-K-001 面,水分子中的氢原子H1 与表面氧原子O3的布居值为0,键长较大,说明并无成键作用,而氢原子H2与表面氧原子O3布居值较大,达到了0.08,成键作用较为明显,形成了一条键长为0.180 nm 的氢键。在None-K-001面,水分子中的氢原子(H6、H7)与表面氧原子(O8、O9)之间的布居值分别是0 与0.01,前者未成键,后者数值较小,形成了键长为0.231 nm 的氢键,键能与键长都较在IN-K-001 面形成的氢键弱。是因为存在钾离子的影响,虽然水分子中的O 原子与钾离子之间的布居值为0,并无成键作用,但是受钾离子的静电作用影响,水分子中的氧原子向钾离子附近靠近。
2.3 吸附后表面静电势分析
见图3、4,在未发生吸附前,伊利石表面的静电势极值点都分布在电负性较大的表面O 原子附近,主要因为表面O 原子的孤对电子对静电势有较大的负贡献,这说明表面O 原子相较于表面Si 原子更容易与水分子形成以静电相互作用为主的吸附。而由于钾离子的存在,IN-K-001 面的极大值点数量比None-K-001 面的极大值点数少。
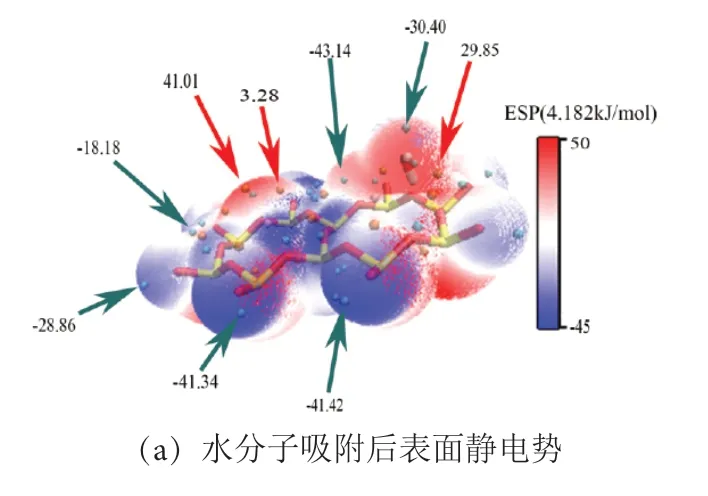
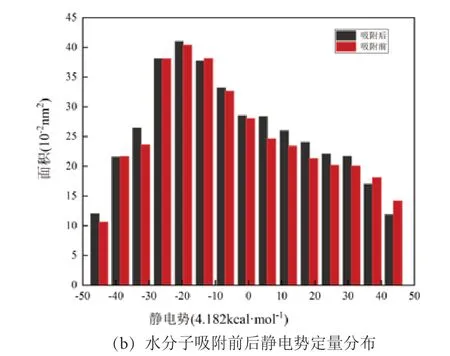
图9 IN-K-001 面水分子吸附后静电势图与定量分布Fig .9 Electrostatic potential map and quantitative distribution diagram of IN-K-001 surface water molecule adsorption
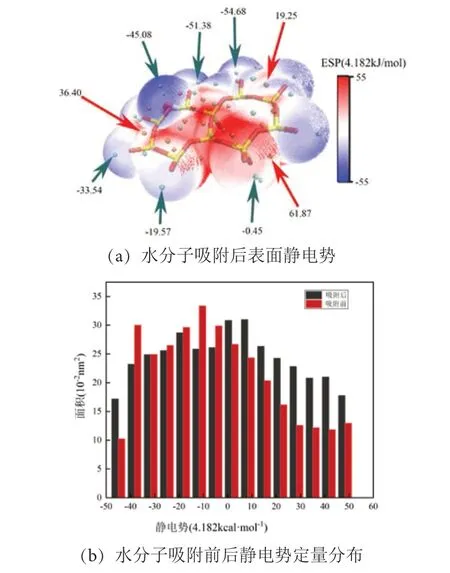
图10 None-K-001 面水分子吸附后静电势图与定量分布Fig .10 Electrostatic potential map and quantitative distribution diagram of No-K-001surface water molecule adsorption
见图9、图10,在水分子吸附后,伊利石001面和001 面的静电势极值点数量增多,水分子吸附部分红色区域均呈增大趋势,而未吸附部分静电势变化不大。见图10(b),在None-K-001 面,静电势为正值区域, 167.28~209.10 kJ/mol 区域面积增加最多,而在IN-K-001 面,增长趋势并不大,这可能因为水分子本身是一个极性分子,在吸附之后,钾离子会与水分子中的氧原子产生弱相互作用,从而减缓了整体的静电势增加。
2.4 电子态密度与电荷分析
电子态密度是能带结构的一个可视化结果,能够反映单位能量间隔内的电子态数目,其中处于EF(费米能级)附近的电子最为活跃,所以可以通过分析各原子的电子态密度,了解各原子的分波成键情况。

图11 IN-K-001 面电子态密度Fig .11 IN-K-001 surface electronic density
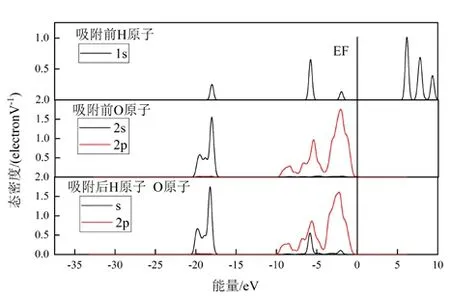
图12 None-K-001 面电子态密度Fig .12 None-K-001surface electronic density
见图11、12,在水分子吸附之前,水分子中H 原子的1s 轨道处在EF 附近,态密度较高,具有较大的反应活性,而在水分子吸附之后,水分子中H 原子与表面O 原子的处在EF 附近的电子态密度都降低(主要是表面氧原子的2p 轨道),说明吸附完成后整体的能量降低,体系更加稳定,也印证了静电势的分析。
见图11,在-12.4 eV~-5 eV 附近,H 原子的1 s轨道与O 原子的2 p 轨道存在较多的重叠,具有较强的成键作用,而在2.3 eV~5 eV 范围内,则存在着反键作用,但是作用强度低于成键效果。见图12,在-7.2 eV~-5.3 eV 附近存在着成键作用,形成了较弱的氢键,有利于水分子的吸附。

表3 水分子在IN-K-001 面吸附前后原子的mulliken电荷分析Table 3 Analysis of mulliken charge of atoms before and after adsorption of water molecules on IN-K-001 surface
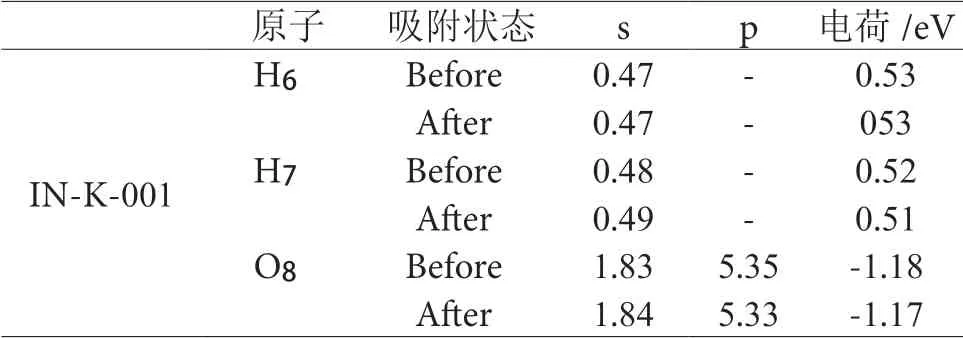
表4 水分子在None-K-001 面吸附前后原子的mulliken电荷分析Table 4 mulliken charge analysis of atoms before and after adsorption of water molecules on the None-K-001surface
表3、4 可知,当水分子吸附到伊利石表面时,成键处氧原子荷负电荷均减少,相应的与之吸附的H 原子荷正电荷数也减少了,结合电子态密度分析可知,并未形成化学键,没有产生电子的转移,而仅仅是形成了电子的聚集。
3 结 论
(1)通过模拟水分子在伊利石IN-K-001 面与None-K-001 面的吸附,确定了水分子在伊利石表面主要是以氢键为主的物理吸附。
(2)伊利石表面静电势负值区域面积大于静电势为正的区域,极小值点个数大于极大值点个数,氧原子附近存在着大量的极值点,这有利于水分子的吸附。水分子吸附后,伊利石表面静电势值较大的红色区域面积增大,吸附活性位点增加。
(3)水分子在伊利石表面稳定吸附后,水分子中的氢原子与表面氧原子之间发生了电子的聚集,电子态密度整体降低, 整个体系的能量降低,导致水分子的吸附更加稳定。
